家族性淀粉样变多发性神经病(FAP)的影像学诊断
时间:2022-12-09 17:59:40 热度:37.1℃ 作者:网络
家族性淀粉样变多发性神经病(familial amyloid polyneuropathy,FAP)为一罕见型常染色体显性遗传疾病,与突变型甲状腺运载蛋白 (transthyretin,TTR) 基因的突变相关。患者一般40-60岁之间发病,临床上以进行性的周围神经、自主神经病变及不同程度的内脏器官淀粉样蛋白质沉积为特征,病人一般于发病后9-13年死亡,预后极差。FAP的病理基础即是甲状腺运载蛋白基因变异和淀粉样蛋白质广泛沉积。
临床表现
患者最主要的临床表现为因淀粉样蛋白质广泛沉积于神经系统所引起的进行性的神经症状,如运动、感觉多神经病症状,自主神经症状如胃肠道症状、神经源性膀胱、直立性低血压、无汗、性功能障碍、心交感神经功能障碍等。患者初始症状为疼痛性外周感觉运动多神经病症状,首先发病于下肢末梢神经,随后表现为上肢,病程后期,视觉、胃肠道、肾脏及心脏皆可受累。心肌淀粉样蛋白质沉积可引起限制性心肌病。
影像学诊断特征
颅后窝和脊髓软脑膜的弥漫性线样强化是FAP累计神经系统的重要特征,心脏受累表现为左心室肥大,心内膜下的延迟期强化。
治疗
原位肝移植可消除肝脏来源的突变型甲状腺运载蛋白淀粉样沉积物,是目前唯一可能的对FAP具有疗效的方法。原位肝移植有令人满意的疗效报道,包括改善自主神经功能、周围神经功能,以及减少内脏淀粉样蛋白的沉积。
患者
52岁女性FAP患者,尿失禁及膀胱不完全排空7月余

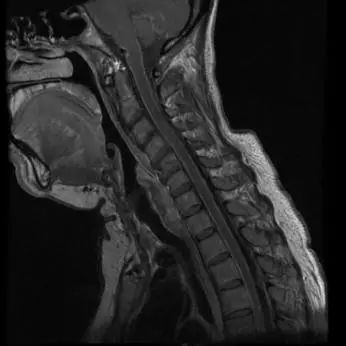
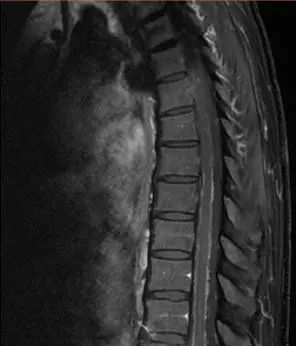

 颅脑轴位、颈髓、胸髓矢状位强化T1WI图像可见脑干、小脑及脊髓软脑膜的弥漫性线样强化,肾脏水平轴位T2WI可见双侧肾盂积水,短轴电影图像可见左心室壁向心性肥厚,T1WI延迟扫描可见前壁的心内膜下强化。
颅脑轴位、颈髓、胸髓矢状位强化T1WI图像可见脑干、小脑及脊髓软脑膜的弥漫性线样强化,肾脏水平轴位T2WI可见双侧肾盂积水,短轴电影图像可见左心室壁向心性肥厚,T1WI延迟扫描可见前壁的心内膜下强化。

