【Ther Adv Hematol】综述:MDS的免疫失调和靶向治疗
时间:2023-09-13 14:01:59 热度:37.1℃ 作者:网络
骨髓增生异常综合征
骨髓增生异常综合征(MDS)是一种克隆性干细胞疾病,以无效造血、形态发育不良和不同程度的血细胞减少为特征,转化为急性髓系白血病(AML)的风险较高。去甲基化异常是MDS的关键机制之一,除表观遗传学异常外,免疫失调也在MDS的发生、发展中起着关键作用。许多研究已阐明免疫系统的不当激活是MDS发病的重要因素,而突变的造血细胞逃避免疫监视可能在高危MDS和进展为AML的生物学中起单独作用。
近年来研究发现,对于骨髓微环境中细胞因子分泌异常的MDS患者,造血微环境中先天和适应性免疫信号均被激活。同时,免疫失调在MDS的发生和进展中起重要作用,尤其是骨髓微环境(包括造血和基质细胞)的破坏。
华中科技大学同济医学院附属同济医院张义成和隗佳教授团队近日于《Therapeutic Advances in Hematology》发表综述,旨在探索免疫细胞、免疫微环境和细胞因子在MDS发病机制中的作用,还讨论了几种不同类别的免疫治疗方法,包括靶向T细胞、直接抑制炎性细胞因子、再利用细胞毒性细胞(repurposing cytotoxic cells)和过继细胞疗法,以更好地理解MDS治疗新手段的开发。本文通讯作者为张义成和隗佳教授,第一作者为张晓颖博士。

免疫细胞
在MDS中,骨髓微环境中的免疫细胞发生改变,尤其是T细胞、NK细胞、巨噬细胞、髓源性抑制细胞(MDSC)和B细胞。许多研究强烈表明,这些免疫细胞数量和功能的改变与MDS进展相关,因此,了解其功能障碍机制对于开发MDS的新靶向治疗至关重要。
T细胞
T细胞的功能障碍在低危MDS的细胞凋亡中起重要作用。一项研究表明,去甲基化药物(HMA)阿扎胞苷可通过诱导肿瘤-睾丸抗原(cancer-testis antigens)上调来增强T细胞对肿瘤睾丸抗原的应答,它是肿瘤监测的基本部分。相反,临床试验表明,在免疫抑制治疗(IST)后发生AML进展的风险类似或较低。MDS肿瘤细胞可通过多种机制逃避肿瘤监测,包括T细胞和细胞因子表达功能障碍以及骨髓基质变化。程序性死亡受体1(PD-1)是一种T细胞表面共抑制受体,可结合程序性死亡配体1/2(PD-L1/PD-L2),防止免疫过度激活。PD-1与PD-L1结合后可破坏T细胞受体(TCR)下游的一系列信号通路,如PI3K/AKT、RAS-ERK1/2和PKC信号通路,从而促进效应T细胞凋亡,抑制细胞增殖和细胞因子分泌(见图1)。但PD-1/PD-L1信号的这种保护功能也可以维持免疫抑制的肿瘤微环境,促进肿瘤细胞增殖。Kondo等人揭示PD-L1仅在原始细胞≥5%的患者中观察到,发现其高表达水平与MDS中高危IPSS评分有关。

CD8+ T细胞
体外研究表明,T细胞在抑制恶性和非恶性造血细胞的生长中发挥作用,并可能由CD8+ T细胞介导,其靶向造血前体上的MHCI类分子。在MDS中,CD8+ T细胞具有直接细胞毒性,产生肿瘤坏死因子α(TNF-α)、白细胞介素6(IL-6)、IL-1Ra、CCL3、CCL4、FAS-L和TRAIL等细胞因子,具有明显的特征。CD8+ T细胞还表现出与T细胞衰竭相关的CD39标志物,表达CD39的T细胞可能通过抑制T细胞活化来促进sAML中的抑制性免疫微环境。
调节性T细胞
调节性T细胞(Tregs)最初被发现是自身免疫的关键免疫调节剂,可通过抑制自身反应性T细胞维持自身耐受。低危MDS中无效造血和骨髓衰竭与免疫紊乱和自身免疫有关,而高危MDS则表现为恶性肿瘤细胞克隆性扩增和免疫逃逸。由于CXCR4的下调,Tregs在MDS早期功能失调,严重影响Tregs通过CXCL12/CXCR4轴的骨髓归巢。许多研究表明,有效抑制局部免疫反应可以促进Tregs选择性迁移到炎症部位,并通过改变Tregs的归巢受体而保留。然而在晚期MDS中,全身和局部Tregs均维持功能和迁移能力。研究认为,Treg扩增可能是由肿瘤相关抗原驱动的,因为Treg克隆是由前白血病克隆和大量肿瘤抗原生长失控导致的,因而肿瘤特异性Tregs可以有效抑制肿瘤相关抗原的特异性免疫反应。Treg抑制的缺乏和骨髓转运失调可在早期MDS的发生发展中起基础作用,而Treg活性的增加可促进晚期疾病白血病克隆的进展。此外,Treg亚型也可能发生转化;一项研究表明,高危MDS患者的一个子集表现出从中央记忆Treg细胞(TregCM)到效应记忆Treg细胞(TregEM)的显著转变。
NK细胞
NK细胞通过分泌细胞因子并通过其溶细胞活性,在宿主防御恶变中发挥重要作用。在高危MDS患者中观察到NK细胞数量减少,导致进一步的克隆演变。然而在低危MDS中,NK细胞似乎对克隆的MDS前体具有细胞毒性,从而抑制进展。除数量变化外,MDS中NK活化受体的表达也显著降低,具有非细胞毒性表型(CD56bright)的NK细胞增加,在疾病早期和晚期均起免疫调节作用,从而并为包括MDS在内的髓系恶性肿瘤的治疗提供机会。
巨噬细胞
巨噬细胞也可能参与MDS的进展。最近的研究表明,巨噬细胞对粒细胞/单核祖细胞的吞噬作用增加可能导致低危MDS患者骨髓中粒细胞/单核祖细胞群周期性和特异性丢失。这一失调的吞噬作用被认为是由靶细胞表面的钙网蛋白和巨噬细胞上的低密度脂蛋白受体相关蛋白(LRP1)受体之间的相互作用所控制。巨噬细胞也可介导血管生成,其在高危MDS中升高。此外,高危MDS巨噬细胞具有低IL-12表达、高IL-10表达、低肿瘤杀伤活性、促进组织重塑和血管生成等特征,这些特征是M2相关的特征。
髓源性抑制细胞(myeloid-derived suppressor cells,MDSC)
MDSC是由趋化因子募集并调节免疫抑制的不成熟髓系细胞的异质性群体,在MDS中提供免疫抑制信号。研究表明MDSC可通过抑制细胞毒性T细胞干扰免疫,而促炎性S100A9与CD33的相互作用促进MDSC扩增。一项研究发现,S100A9和CD33形成一个功能性配体/受体对,招募CD33的免疫受体酪氨酸抑制基序(ITIM)的组分,诱导抑制性细胞因子IL-10和TGF-β的分泌。除免疫调节作用外,MDS中MDSC分泌的炎症介质还可直接破坏红细胞生成,促进疾病进展。
B细胞
多项研究证明,许多早期MDS患者表现为B细胞祖细胞室的异常。早期MDS的一个特征可能为,主要在B细胞祖细胞中表达的基因表达减少。与骨髓正常者相比,MDS患者骨髓CD19+细胞凋亡水平明显。此外与正常细胞相比,5q综合征患者的B细胞或其前体数量显著减少,pro-B(CD34+19+)细胞频率也显著降低。
基质微环境
MDS是整个骨髓的功能性疾病,包括造血细胞和间充质成分。对MDS患者骨髓功能的研究表明,造血细胞与基质细胞有密切关系。人骨髓成纤维细胞的集落形成单位(CFU-F)分析表明,与从健康对照中纯化的骨髓间充质干细胞(MSC)相比,MDS患者的骨髓间充质干细胞的CFU-F计数减少。此外与健康对照样本相比,MDS样本的MSC在培养物中未保持高传代。Dicer1缺陷小鼠模型证明,功能失调的基质环境可能启动骨髓发育不良。另一项研究证明,MDS的小鼠模型可以更有效地移植到老年受体小鼠体内,而非移植到年轻受体小鼠体内,表明老年骨髓基质更有利于MDS的发生。在一个MDS小鼠模型中,MSC中WNT/β-catenin信号的增加和MSC来源的成骨细胞中β-catenin的激活导致AML的发生,表明骨髓基质的WNT信号也可促进MDS的进展。此外,在小鼠癌症模型中部分MSC遗传通路(WNT/β-catenin、Jagged-1、促炎基因、miR-155)也与人类临床结局相关。转录组分析揭示了细胞应激和炎症相关分泌因子上调的MDS患者骨髓基质细胞的转录特征。由此可见,MDS的某些方面可能是由骨髓间充质干细胞所驱动,而其他方面则可能是MDS进展和转化为白血病的机制。
炎性和异常细胞因子的生成
近年来发现MDS的主要致病因素是恶性克隆和天然免疫活化异常,以及骨髓微环境中的促炎信号转导。Toll样受体(TLR)信号参与免疫应答,但在MDS中,TLR及其下游效应器存在异常激活。研究表明,低剂量脂多糖(LPS)可激活TLR信号,改变造血。此外,一项使用转基因小鼠模型的研究证明,过表达S100A9也会诱导血细胞减少和发育不良造血。此外,S100A9介导的节点样受体蛋白3(NLRP3)的炎症激活可导致焦亡细胞死亡,它是疾病许多典型特征的基础。该通路以及伴随的其他风险相关分子模式的释放,可扩增MDSC,从而创造一个正反馈过程,可放大炎症体(inflammatory body)活化。在炎症体家族中,NLRP3与MDS细胞的焦亡有关;此外在MDS中,焦亡相关基因转录和炎症体组装显著上调。不同功能类别的体细胞基因突变可导致NLRP3具有共同的表型,包括活性氧的过度产生、Wnt/β-catenin诱导的增殖、阳离子流诱导的细胞肿胀、和caspase-1活化。尽管这些发现与观察到的MDS细胞的竞争力相矛盾,但NLRP3作为MDS扩增驱动因子之间的关系有待进一步探索。
高迁移率族蛋白B1(HMGB1)是一种核蛋白,参与炎症状态下染色质折叠、转录和信号传导;可由坏死细胞被动脱落,也可由单个核细胞主动释放,从而进一步放大炎症。异常的炎症信号诱导骨髓祖细胞凋亡、NLRP3炎症体激活和焦亡,从而可能通过干扰血红蛋白稳态和EPO信号诱导贫血。循环HMGB1在MDS中增加,但在其他骨髓衰竭综合征中没有增加,这进一步表明HMGB1参与MDS的免疫发病机制。HMGB1抑制剂和中性粒细胞弹性蛋白酶已与阿扎胞苷联合用于减少异常(但不健康)MDS CFU的体外扩增。研究还显示,抑制HMGB1可降低LR-MDS细胞中TLR和NF-κB的表达;因此它可能作为MDS的治疗靶点。近年来的研究表明,阿司匹林可能通过抑制HMGB1的活性而发挥减轻炎症的作用,因此阿司匹林减轻MDS炎症的可能有益作用值得探讨。
异常的细胞因子在MDS的免疫失调中起着复杂而重要的作用,在MDS患者的骨髓样本中发现许多细胞因子和生长因子的水平异常。此外,在MDS患者的骨髓和血清中,TNF-α水平升高尤其与多种效应相关,如细胞凋亡增加、骨髓细胞数量增加、造血抑制以及下游信号通路和转录因子激活。细胞因子在调节细胞间相互作用中起着至关重要的作用,免疫细胞的行为和功能也受到与细胞因子相互作用的调节。例如,辅助性T细胞17(Th17)T淋巴细胞通过产生IL-17而发挥作用,IL-17是一种细胞因子,可以反过来激活巨噬细胞和DC而产生额外的促炎细胞因子。研究表明,低危MDS中IL-17水平升高,且可能在诱导细胞凋亡中起作用。
炎症和免疫失调在MDS的启动和进展中至关重要。MDS和慢性粒单核细胞白血病(CMML)通常与自身免疫性疾病(AD)和免疫系统的炎症反应相关。应考虑血细胞减少背景下发生的AD与MDS相关,尤其是在老年患者中。此外,血细胞减少似乎是一些MDS患者复杂的自身反应性免疫活性的结果,可能对IST有反应。炎性细胞因子(如TNF-α和干扰素)的释放增加,可触发骨髓前体细胞凋亡,导致血细胞减少。免疫细胞(包括细胞毒性Treg、Th17和NK细胞)功能受损也可预测IST应答、AD结局和发生。空泡(Vacuoles)、E1酶(E1Enzyme)、X-连锁(X-Linked)、自体炎症(Autoinflammatory)和体细胞(Somatic)突变综合征是一种新描述的成人发作性炎症综合征,与MDS和AD重叠。突变导致UBA1典型胞浆亚型的丢失,泛素化减少,天然免疫途径和全身炎症的激活。既往研究证明,抗炎药物无法改善任何研究对象的VEXAS综合征,但所有患者均为高剂量糖皮质激素依赖性。一项纳入11例确诊VEXAS综合征的MDS患者的研究中,阿扎胞苷治疗的缓解率为46%。值得注意的是,与MDS相关的克隆性T细胞大颗粒淋巴细胞(T-LGL)增殖并不少见。在一项更大规模的研究中,Huh等人描述了9例同时患有MDS和T-LGL的患者,并提出了两者之间的病因学关系,而非简单巧合。一项比较有和无T-LGL增殖的MDS患者的研究发现,MDS患者的T-LGL增殖可能与骨髓细胞减少和谱系发育不全有关。此外,自身反应性T细胞可能抑制造血功能,导致T-LGL和部分MDS患者血细胞减少,可能导致T-LGL/MDS的发生。IST的发生可能有利于T-LGL增殖的MDS患者中T-LGL细胞的消除。
靶向MDS免疫系统的治疗
MDS具有高度异质性,从而对开发新型治疗方法提出了独特的挑战。单纯纠正免疫微环境不足以治疗MDS,因此可能需要免疫疗法联合其他药物来最终阻止疾病进展。由于MDS不同分期的免疫特征不尽相同,多项试验探讨了免疫调节在低危和高危MDS患者中的潜在作用,在表1中简要综述。

免疫抑制治疗
靶向T细胞的抗胸腺细胞球蛋白(ATG)和环孢素(CSA)对部分MDS患者有效,尤其是病态造血患者。研究表明,两种治疗的缓解率差异很大,联合治疗并不优于单药治疗。在一项II期研究中,25例输血依赖性MDS患者接受了一个疗程的ATG治疗,部分患者的造血功能恢复,尤其是难治性贫血患者,且耐受性良好。此外,一项ATG和CSA免疫抑制治疗的单中心研究结果表明,早期MDS患者的缓解率与其他标准治疗相似,但晚期MDS患者对IST的反应较差。一项大型、多中心国际队列研究回顾性评估了207例接受IST的MDS患者,ORR为48.8%,表明用于细胞减少性骨髓患者的首选IST方案为马ATG联合CSA。一项开放标签随机III期研究也表明,ATG和CSA治疗与血液学缓解相关,对TFS和OS无明显影响,其中病态造血MDS的ORR更高,为50%。多项研究也证明IST治疗显著有利于生存率,但报告的结果相互矛盾;免疫抑制药物在MDS中仍存在争议。IST与MDS的相关性取决于特定亚型的骨髓失败是否具有自身免疫成分。一些研究表明,它可能使某些具有特定特征的MDS患者获益:病态造血、HLA-DR15、8三体综合征、年轻(<60岁)、无体细胞突变和低输血负荷。
直接抑制炎性细胞因子
抗TNF-α治疗是早期MDS中用于靶向细胞因子水平异常的主要策略之一。一些关于依那西普和英夫利昔单抗的研究证明了早期活性,然而一项II期试验也证明了低活性和低反应。与其他药物联合使用的研究也很一般。依那西普联合阿扎胞苷治疗3个月后的总缓解率为72%,但本研究中用于评估缓解的标准对阿扎胞苷单药治疗至关重要。不幸的是,TNF-α抑制剂并不像预期的那样成功,目前还没有被用作MDS的标准治疗。IL-6作为参与MDS发病的重要细胞因子,也被用于MDS的治疗,但结果也一直较差。一项双盲、II期研究评估了司妥昔单抗,但由于在减少红细胞(RBC)输注方面缺乏疗效,该研究提前终止。然而,靶向细胞因子信号的新策略仍具有一定的治疗潜力,例如罗特西普在II期研究中表现出了可喜的结果,能够结合转化生长因子β超家族配体来减少SMAD2和SMAD3信号,改善红细胞生成。一项关于罗特西普治疗MDS的安慰剂对照、双盲、III期试验显示,38%的患者可在8周或更长时间内摆脱输血依赖。
再利用细胞毒性细胞
目前尚不清楚髓系恶性肿瘤中靶向转化细胞的细胞毒性免疫反应的作用,包括刺激内源性系统和重组淋巴源性细胞以突变细胞为靶点。随着免疫检查点抑制剂在实体瘤临床治疗中的成功应用,免疫检查点阻断疗法的概念已被应用于血液肿瘤。阻断免疫检查点可能是治疗晚期MDS的有效合理策略,包括抑制PD-1/PD-L1通路在MDS免疫逃逸和细胞毒性T细胞衰竭中的作用。帕博利珠单抗可阻断PD-1与其PD-L1配体的相互作用,然而在28例HMA治疗失败的MDS患者中,24周后帕博利珠单抗单药治疗的ORR仅为4%,OS率为49%。临床前研究与临床试验矛盾的原因尚不清楚,但骨髓免疫微环境的动态变化可能是关键。另一项II期试验在37例IPSS中危-1或更高危MDS患者中,评估了帕博利珠单抗和AZA的协同效应,结果HMA初治队列(n=17)的ORR为76%,HMA失败队列(n=20)的ORR为25%,CR分别为18%和5%。此外,在中位随访12.8个月后,HMA初治队列未达到中位总生存期(mOS),而HMA治疗失败队列为5.8个月。这些结果表明,HMA和PD-1/PD-L1抑制剂具有潜在的协同效应,但基于PD-1/PD-L1抑制剂治疗MDS仍存在明显挑战。
过继细胞疗法
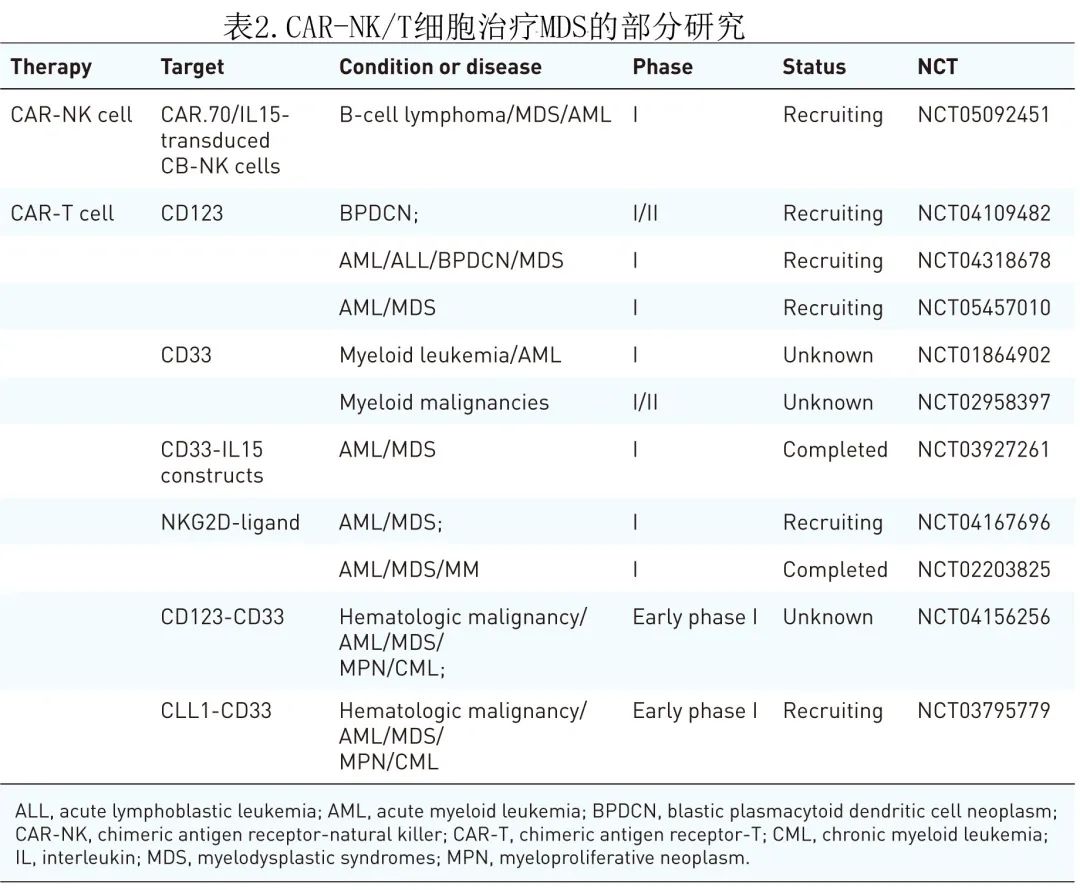
工程化NK细胞毒性治疗病态造血克隆是一种新的治疗方法,在AML和晚期MDS中表现出一定的积极作用。一项关于NK细胞治疗的试验证明,高危MDS患者对治疗有反应,从而支持使用单倍体相合NK细胞输注作为难治性患者HSCT的桥接治疗。在单倍体相合HCT后的高危AML和MDS患者中开展的一项II期随机试验也证明了单倍体相合HCT后NK细胞治疗在减少疾病进展方面的获益。继成功治疗淋巴瘤后,多项试验也评估了CAR-T细胞在MDS和其他晚期髓系肿瘤中的作用。CAR-T细胞需要对恶性细胞具有一定程度的特异性,以确保有健康的祖细胞及时重新填充骨髓,从而避免并发症。已经开发了多种靶向CD123的CAR产品,使用起源于正常祖细胞的高危MDS干细胞。一项首次人体I期研究(NCT02159495)纳入了40例受试者,评估MB-102(CD123 CAR-T细胞)的抗肿瘤活性和安全性,证实在AML和母细胞性浆细胞样树突状细胞肿瘤(BPDCN)患者中达到完全缓解且无剂量限制性毒性。CD33CAR-T细胞治疗证实,CAR-T细胞输注在1例患者中引起了严重的毒副作用,包括全血细胞减少症加重和血清细胞因子水平升高,CAR-T细胞治疗2周后患者骨髓原始细胞明显减少,但9周后显著疾病进展开始恢复。还对组合靶点进行了评价,如CD123–CD33 cCAR-T细胞(NCT04156256)、CLL1–CD33(NCT03795779)或CD33–IL15结构(NCT03927261)。另一项关于CAR-T细胞的研究发现,这些细胞能够识别NKG2D配体,在AML和MM中没有产生显著的临床活性。此外,目前正在进行抗NKG2D CAR–T细胞(常见于MDS克隆)的I期试验(NCT04167696)。当前CAR-T细胞疗法相关的主要挑战之一是缺乏特异性抗原,许多肿瘤相关抗原也在正常骨髓细胞上表达,对非MDS靶细胞发挥清髓作用。表2中简要报道了目前正在研究的一些CAR-NK/T细胞疗法。
结论和未来展望
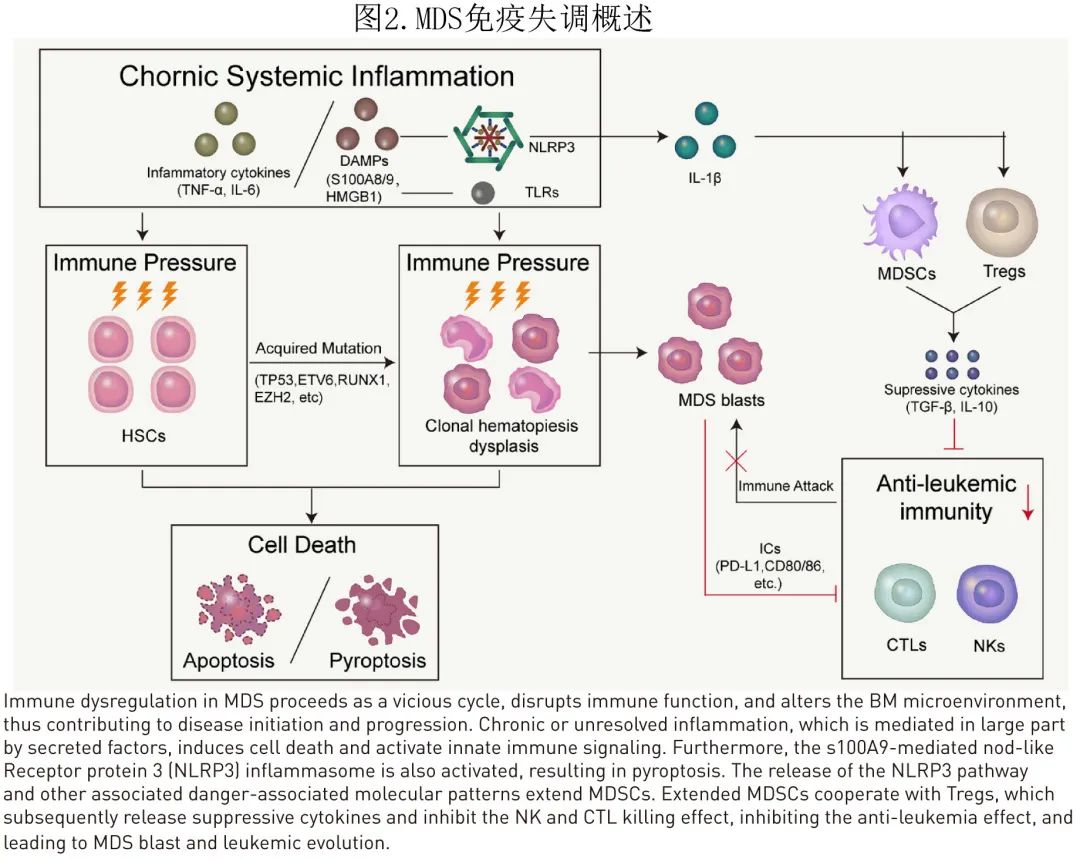
免疫细胞、产生异常细胞因子的炎症信号和基质微环境是MDS疾病表型和临床表现的重要促成因素。图2描述了这些因素的可能机制。随着多组学方法在骨髓微环境中的应用,将进一步更详细地阐明MDS发病机制。MDS的突变和临床异质性是成功治疗MDS的挑战,而改善造血微环境可能促进部分患者造血功能的恢复,抑制疾病进展,因此有必要了解不同疾病阶段炎症和微环境的变化,以便构建靶向治疗,对抗疾病的促炎环境,最终阻止疾病进展。同时考虑到疾病的复杂性,可能需要联合治疗。当下已经做出了重要的努力来寻找治疗免疫系统的方法,包括激活静止的免疫效应细胞和改善异常的炎症微环境。关于可用于评估免疫治疗反应的预测指标的额外工作,包括CAR-T,也是非常有必要的。
参考文献
Xiaoying Zhang , Xingcheng Yang , Ling Ma , Yicheng Zhang, Jia Wei.Immune dysregulation and potential targeted therapy in myelodysplastic syndrome.Ther Adv Hematol . 2023 Aug 2;14:20406207231183330. doi: 10.1177/20406207231183330.
