【协和医学杂志】国内首例FOXN1单倍体不足报告并文献复习
时间:2023-09-26 20:19:37 热度:37.1℃ 作者:网络
叉头盒(FOX)基因家族N1(FOXN1)纯合/杂合突变均可影响胸腺上皮细胞(TECs)发育致使T淋巴细胞成熟障碍,进而导致疾病的发生。由FOXN1杂合突变导致的FOXN1单倍体不足主要表现为婴儿期T淋巴细胞减少,反复感染,伴或不伴先天性无发、甲营养不良等[1]。
本文报道国内首例FOXN1基因杂合突变导致的FOXN1单倍体不足病例,对其临床表现、免疫表型、突变位点和诊疗措施等进行介绍,并进行文献复习,以增进临床医师对此疾病的了解和识别能力。
1 病历资料
患儿为女婴,1岁5个月,因发现贫血1年余,2021年6月24日于北京协和医院儿科门诊就诊。患儿足月顺产(第4胎第2产),出生体质量2350 g,出生后无窒息缺氧史,无病理性黄疸史。生长发育史无异常。按时接种疫苗,接种卡介苗后接种部位出现红肿破溃持续2月余(图1A)。
入院查体:体质量7.8 kg,反应可,贫血貌,毛发稀疏(图1B),反甲(图1C),心肺腹部查体未见异常,四肢关节及肌张力无异常。
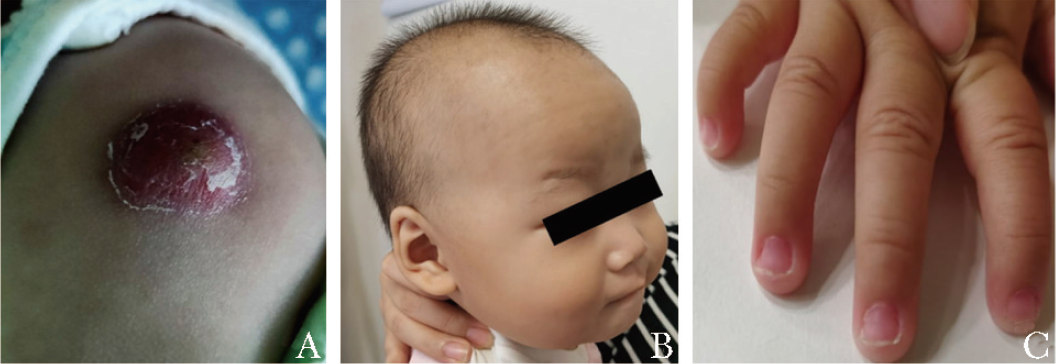
图1 患儿入院查体体征
A.卡介苗接种部位局部反应;B.毛发稀疏;C.反甲
1.1 辅助检查
血常规:红细胞 1.16×1012/L,血红蛋白 45 g/L,平均红细胞体积 113.8 fL,平均红细胞血红蛋白含量38.8 pg,网织红细胞百分比 18.90%,网织红细胞绝对计数219.2,白细胞及血小板大致正常。
尿、便常规均未见异常。
血生化:谷草转氨酶 77.3 U/L,谷丙转氨酶 77.1 U/L,肌酸激酶正常,乳酸脱氢酶819 U/L,总胆红素 29.51 μmol/L,直接胆红素3.47 μmol/L,间接胆红素 26.04 μmol/L,肾功能正常。
感染指标筛查均为阴性。
免疫:免疫球蛋白三项正常,红细胞沉降率 106 mm/h,补体C3 0.53 g/L,C4正常,抗核抗体 1:20,其余自身抗体谱均为阴性。
TB淋巴细胞亚群:总T淋巴细胞1284个/μL,CD4+ T 淋巴细胞195个/μL,CD8+ T淋巴细胞 956个/μL,总B淋巴细胞2021个/μL,NK细胞507个/μL。
凝血功能正常,直接抗人球蛋白试验阳性(4+)。
血涂片:成熟红细胞胞体大小悬殊,可见变形红细胞,可找到球形红细胞。
骨髓涂片:骨髓增生活跃,红系增生旺盛,中幼红比值增高,粒红比值降低。
1.2 基因检测
征得患儿父母同意后,采集患儿及其父母外周血标本,进行全外显子基因组测序,并进一步对检测到的疑似突变位点进行Sanger测序验证。
全外显子组测序发现患儿存在FOXN1基因(NM_003593)杂合移码突变,c.1392_1401delTCCTGGACCC(p.P465Rfs*82)为新生突变(图2),且该位点为致病突变,目前国内尚无报道。
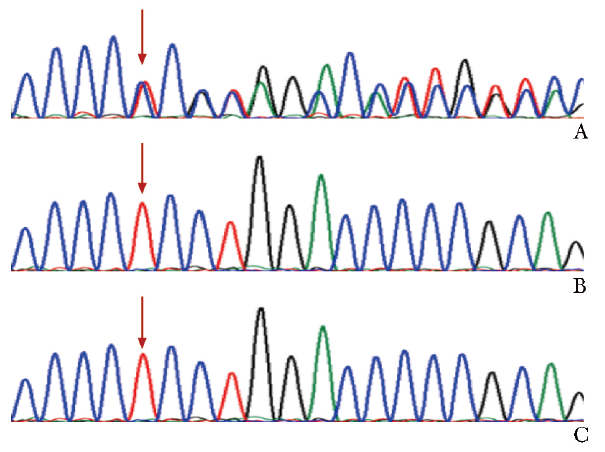
图2 患儿及其父母FOXN1基因Sanger测序图(箭头为突变位点)
A.患儿FOXN1基因存在移码突变c.1392_1401del TCCTGGACCC;B.患儿父亲无该突变;C.患儿母亲无该突变
1.3 TB淋巴细胞亚群分析
采集患儿外周血标本,采用流式细胞术检测外周血TB淋巴细胞各亚群分布情况。
该患儿17个月龄时淋巴细胞绝对数为5263个/μL(参考范围:2980~5950个/μL);总T淋巴细胞3042个/μL(1175~3953个/μL),占总淋巴细胞的57.80%(53.40%~71.91%);CD4+ T淋巴细胞532个/μL(948~2477个/μL),占总T淋巴细胞的17.49%(26.19%~45.48%);初始CD4+ T淋巴细胞58个/μL(530~1837个/μL),占CD4+ T淋巴细胞淋巴10.90%(46.42%~81.20%);记忆CD4+ T淋巴细胞467个/μL(210~850个/μL),占总CD4+ T淋巴细胞的87.78%(7%~20%);CD8+ T淋巴细胞1821个/μL(531~1521个/μL),占总T淋巴细胞的59.86%(16.29%~29.88%);CD4/CD8比值为0.29(1.05~2.53);B淋巴细胞1168个/μL(537.11~1464.39个/μL),占总淋巴细胞的22.19%(13.93%~30.49%);NK细胞1010个/μL(241~978个/μL);占总淋巴细胞的19.19%(6.5%~22.24%)。
1.4 T淋巴细胞抗原受体剪切环、κ-删除重组切除环和T淋巴细胞受体Vβ检测
采集患儿外周血标本制成干血片,提取DNA,通过实时定量聚合酶链式反应(PCR)检测外周血T淋巴细胞抗原受体剪切环(TRECs)和κ-删除重组切除环(κRECs)拷贝数。该患儿TRECs拷贝数为0.35 copies/μL(正常值≥33 copies/μL),提示T淋巴细胞生成缺陷;其κRECs正常。
采集患儿外周血标本,提取RNA并逆转录,通过PCR检测T淋巴细胞受体(T cell receptor,TCR)Vβ多样性。结果显示,患儿TCR Vβ多样性受限(图3),测序结果为寡克隆,而正常人为对称的钟形分布。
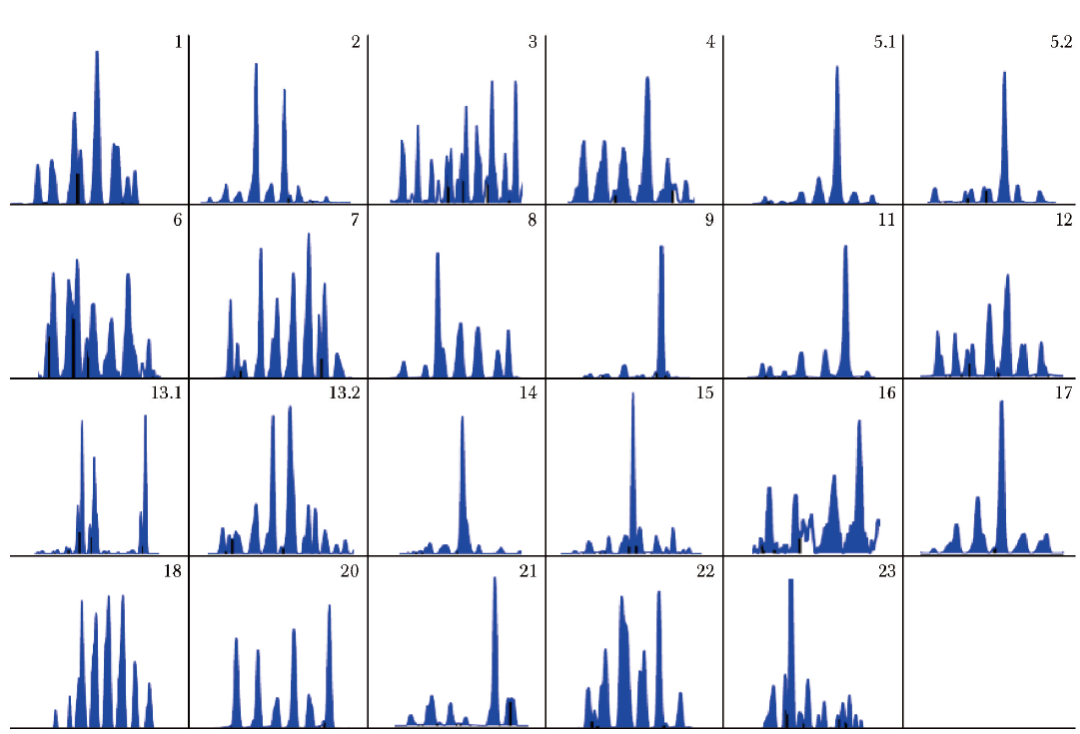
图3 患儿TCR Vβ多样性测序结果
1.5 诊疗经过
2020年12月27日,予患儿静脉输注甲强龙(210 mg×3 d)、免疫球蛋白(7.5 g×1 d)冲击治疗后,输注洗涤红细胞,1周后患儿血红蛋白逐渐回升。之后予利妥昔单抗输注治疗(0.14 g/周×4次),并规律随访。治疗1个月后,患儿出现低丙种球蛋白血症,予定期输注丙种球蛋白(每月200 mg/kg)。患儿血红蛋白恢复并稳定后,停用利妥昔单抗。
随访过程中患儿再次出现血红蛋白降低,于2021年2月开始予以雷帕霉素(1 mg/d)治疗,血红蛋白逐渐恢复正常。雷帕霉素治疗1个月后,患儿出现肝酶进行性升高,考虑为雷帕霉素副作用所致,于2021年9月更改治疗方案为吗替麦考酚酯(0.25 g/d)。后续随访中患儿肝酶持续下降并恢复正常,T淋巴细胞计数逐渐上升,血红蛋白水平稳定。
2 文献复习
以“FOXN1 deficiency”“FOXN1 haploinsuffici-ency”“FOXN1缺陷”“FOXN1单倍体不足”为检索词在PubMed、万方数据知识服务平台和中国知网数据库进行文献检索,检索时间为建库至2021年8月31日,共检索到符合要求的英文文献5篇[1-5],无相关中文文献。
对检索到的相关文献进行复习,包括本例患儿在内共103例患者携带FOXN1杂合突变,其中儿童患者41例。进一步对其突变类型进行分析发现,103例患者共携带26种不同的突变类型,其中移码突变15种(57.7%),错义突变8种(30.8%),无义突变2种(7.7%),剪接位点突变1种(3.8%)(图4)。
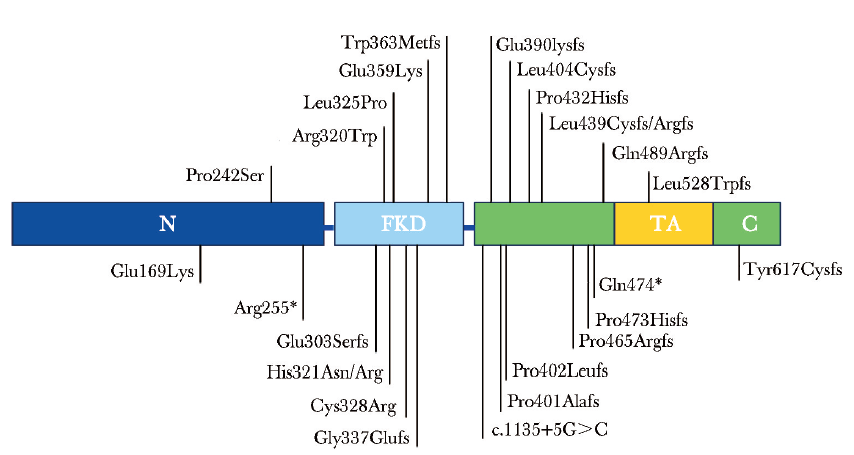
图4 文献报道的FOXN1基因杂合突变位点示意图
临床表型方面,携带FOXN1基因杂合突变患者的临床表现类似于FOXN1纯合缺陷患者,主要表现为反复感染、甲营养不良及毛发发育异常,但此类患者的临床表型存在较大异质性,轻者可无任何临床症状,重者可因反复严重感染而威胁生命。
结合文献及本例患儿的情况分析,所有病例均报告了甲营养情况,其中59例(57.3%)存在甲营养不良,手足指甲均可受累,主要表现为白甲、反甲、管状甲营养不良以及博氏线形成;64例报告了感染情况,其中28例(43.8%)存在反复感染,且儿童患者感染发生率(72%)明显高于成人患者(27.3%),主要表现为各种病毒感染,临床表现以反复上呼吸道感染最为常见,其次是肺炎,部分患者表现为慢性腹泻,其中1例患儿(与本病例携带相同基因突变)还患有EB病毒相关的B细胞淋巴瘤;24例报告了毛发情况,其中12例(50%)出现毛发发育异常,主要表现为头发稀疏,2例无头发生长,4例表现为眉毛发育异常;13例报告了胸腺发育情况,其中10例(76.9%)存在胸腺形态异常(本例患儿胸腺发育无异常),6例为胸腺体积缩小,4例存在胸腺影缺失;6例报告了生长发育情况,其中3例存在生长发育迟缓;6例报告了自身免疫情况,其中4例存在自身免疫现象,1例为银屑病,1例为1型糖尿病,1例为严重免疫性血小板减少症。本例患儿为自身免疫性溶血性贫血。此外,少数患者还存在湿疹、特应性皮炎、Omenn综合征等表现。
免疫表型方面,64例患者评估了淋巴细胞水平,所有患者B淋巴细胞和NK细胞数量均正常;初次检测时41例患儿均存在T淋巴细胞减少,表现为CD4+ 和CD8+ T淋巴细胞同时减少,其中21例患儿出生后进行新生儿筛查时因TRECs较低,进一步检查发现存在T淋巴细胞减少。随着年龄的增长,大部分患儿总T淋巴细胞及CD4+ T淋巴细胞数量可逐渐恢复正常,而CD8+ T淋巴细胞数量随年龄增长改善不明显,绝大部分成人患者总T淋巴细胞及CD4+ T淋巴细胞均在正常范围,但CD8+ T淋巴细胞数量仍低于正常值。进一步对T淋巴细胞亚群分析发现,无论其绝对计数是否正常,此类患者的CD4+和CD8+初始T淋巴细胞始终处于较低水平。此外,13例患儿检测了T淋巴细胞增殖功能,其中4例存在异常,2例在后续随访过程中恢复正常。
治疗方面,根据文献报道,目前针对FOXN1单倍体不足的患者,主要采取保守对症支持治疗,对于具有低丙种球蛋白血症的患儿可定期予以免疫球蛋白替代治疗,对于感染风险较大的患儿可予以复方新诺明或阿昔洛韦预防感染。5例患儿因T淋巴细胞显著缺乏导致严重反复感染而进行了造血干细胞移植,其中1例于移植2年后死于严重的移植物抗宿主反应。
3 讨论
FOXN1基因缺陷是重症联合免疫缺陷(SCID)中较少见的一种常染色体隐性遗传病,也被称为“nude SCID”,最早于1966年提出,用于描述具有“裸”表型的小鼠[6-7],直到1996年才首次报道具有类似表型的人类患者[8]。
目前,文献报道的FOXN1纯合突变患者不足20例,此类疾病除具有典型的T淋巴细胞缺陷导致的SCID表型外,还伴有先天性无发和甲营养不良[2,9-14]。若不及时诊治,大部分患者可因反复严重感染而死亡。
近年来,随着新生儿筛查在国外的推广和普及,一些具有低TRECs但表现不典型的杂合突变患者逐渐被发现[1]。初步研究表明,此类患者因FOXN1基因杂合失功能突变引起FOXN1基因剂量不足而导致疾病发生,表现为常染色体显性遗传模式[1]。
2019年,国际免疫学联合会发布的免疫出生错误最新分类中将FOXN1单倍体不足首次纳入具有相关综合征或综合征特征的联合免疫缺陷病分类中[15]。其临床表现与经典的FOXN1缺陷有所不同,可表现为因T淋巴细胞降低而导致的反复呼吸道感染、皮肤病变(包括湿疹、皮炎)、毛发异常及甲营养不良。但由于突变位点的不同,临床表现和病情严重程度存在较大差异,轻者可无任何临床症状,重者可因反复感染而危及生命,但整体严重程度低于FOXN1缺陷患者[1]。
FOXN1是FOX转录因子家族的一员,其编码基因位于人类染色体17q11.2[1],包含两个重要结构:一个位于中间位置的Forkhead DNA结合结构域,可与含有GAa/cGC结构的DNA序列相互作用,转录激活近500个基因,包括角蛋白、蛋白酶体成分和细胞表面蛋白相关基因[16];另一个位于C端的反式激活结构域(TA),为激活靶基因所必须的结构域(图4)[17]。
FOXN1特异性表达于TECs和角化细胞,是TECs发育过程中的关键调控因子,进而对胸腺形成和T淋巴细胞的发育、分化及成熟产生重要影响[18-20]。FOXN1功能缺失可导致小鼠胸腺体积显著缩小,胸腺组织内缺乏TECs生长,不能形成有功能的髓质TECs和皮质TECs,因此无法为T淋巴细胞提供正常的发育微环境,导致其发育受阻;角化功能异常将影响毛囊发育,致使毛发不能正常生长,导致小鼠出现“裸”表型[4]。
此外,FOXN1可通过促进诱导早期淋巴祖细胞向胸腺迁移的相关基因(如Ccl25、Cxcl12、Dl4和Scf)表达以参与T淋巴细胞发育。研究发现,FOXN1可直接调控参与抗原加工和胸腺细胞选择过程的基因表达,如Psmb11、Prss16和Cd83[21]。FOXN1杂合突变小鼠的TECs内上述基因表达均显著降低[1]。另一方面,FOXN1在TECs的稳定表达在个体生命周期中对维持胸腺功能、避免胸腺过早退化也发挥重要作用[22-23]。
通过文献复习可知,FOXN1单倍体不足患者其突变类型大多为移码突变,该突变可导致翻译提前终止,产生截断蛋白,降低转录因子的活性,且突变主要位于Forkhead区和可影响反式激活结构域的区域[1],这可能也是杂合突变导致疾病发生的一个重要原因。因此,此类患儿临床表现的巨大差异可能来源于不同突变位点对FOXN1活性影响的严重程度。
另一方面,FOXN1不同区域对TECs和角质细胞的调控差异可能也是造成临床表现变化较大的一个因素。研究表明,位于该基因1号外显子上一个长1.6 kb的高度保守区域功能缺失可导致胸腺发育完全受损,但角质细胞功能却不受影响[24]。
此外,研究发现婴幼儿患者的症状明显重于成人患者,且大部分成人患者基本无症状,虽然部分成人患者儿童期有严重反复感染发生,但随着年龄的增长,症状均可逐步缓解[1,5]。
一方面,可能与成人患者T淋巴细胞数量增加有关,本例患儿在随访过程中亦观察到了T淋巴细胞数量逐渐恢复正常。但T淋巴细胞数量随年龄增加逐渐恢复的机制目前尚不清楚,推测可能为T淋巴细胞稳态增殖的结果。虽然T淋巴细胞数量恢复正常,但由于胸腺功能的不可逆丧失,初始T淋巴细胞始终不能恢复正常,增加的T淋巴细胞主要为记忆T淋巴细胞,且研究发现此类T淋巴细胞受体主要为寡克隆[25]。
对本例患儿TCR Vβ检测结果进行分析发现,其TCR亦表现为寡克隆状态。由于胸腺功能缺陷,外周T淋巴细胞未经胸腺的阳性和阴性选择,因此存在较多自身反应性T淋巴细胞,更易发生自身免疫反应。文献报道的病例中存在有合并银屑病、免疫性血小板减少和1型糖尿病患者[5],本例患儿则表现为自身免疫性溶血性贫血。
另一方面,FOXN1剂量效应的时间依赖性可能随基因启动子区甲基化的改变而改变[26]。随着患儿年龄的增长,启动子区甲基化水平升高,导致生理状态下FOXN1基因的转录活性逐渐下降,因此这也可能是剂量效应在成人患者中体现不明显的原因之一。
本例患儿携带的突变基因与Giardino等[5]所报道的病例家系相同,该突变基因可导致TA结构域不能正常转录。该家系中3例患儿及其父亲、祖母均携带此突变基因。3例患儿均表现为反复严重感染,其中1例在随访过程中发生了EB病毒相关的淋巴瘤,1例患儿因反复感染而进行了骨髓移植,于2年后死于移植物抗宿主反应。
患儿父亲幼时也出现反复严重感染,但随着年龄的增长感染发生次数逐渐减少,而患儿祖母除患有1型糖尿病外无其他任何症状。毛发异常也仅表现在3例患儿及其父亲身上,且仅局限于眉毛异常。本例患儿除卡介苗接种后出现轻微异常外,无其他任何反复感染情况,且以自身免疫性溶血性贫血为突出表现,伴有不典型的毛发发育异常和甲营养不良,进一步证实该病具有极大的临床异质性。造成此种现象的可能原因为存在有非编码区相关调控序列的异常或显性负效应[5]。
目前,针对FOXN1缺陷尚无针对性治疗方案,因患者T淋巴细胞功能异常是由TECs发育不良所致,属于骨髓外因素,因此通过常规的造血干细胞移植并不能根治此类疾病。根据既往报道,通过造血干细胞移植后患者总T淋巴细胞数量可有一定程度的恢复,但是初始T淋巴细胞始终无法恢复正常。虽有研究指出,FOXN1缺陷患者接受骨髓移植可降低感染的发生率,但主要归功于骨髓移植物中存在具有增殖能力的供体成熟T淋巴细胞[27]。
胸腺移植可能是唯一治愈此类疾病的手段,可使患者的免疫功能得以重建,各T淋巴细胞亚群及TCR Vβ多样性得以恢复,进一步使机体免疫功能趋于正常[9]。但对于杂合突变患者来说,由于其T淋巴细胞减少随年龄的增长逐渐恢复,临床症状逐渐改善,目前主要采取对症支持治疗。
4 小结
本文报道了国内首例FOXN1单倍体不足患者,通过文献复习发现此类疾病患者临床异质性极大,主要表现为可随年龄增长而改善的T淋巴细胞减少,临床可表现为反复感染、毛发发育异常、甲营养不良、自身免疫异常、胸腺发育不良等。本例患儿以反复自身免疫性溶血为主要表现,为一种全新的表型。需注意的是,虽然原发性免疫缺陷病多以早发反复感染为主要表现,但当缺乏典型的临床表现时,仍需警惕一些非典型临床特征,以更好地协助诊断,特别是对于具有综合征表型的免疫缺陷病。
参考文献
[1]Bosticardo M,Yamazaki Y,Cowan J,et al. Heterozygous FOXN1 Variants Cause Low TRECs and Severe T Cell Lymphopenia,Revealing a Crucial Role of FOXN1 in Supporting Early Thymopoiesis[J]. Am J Hum Genet,2019,105:549-561.
[2]Adriani M,Martinez-Mir A,Fusco F,et al. Ancestral founder mutation of the nude (FOXN1) gene in congenital severe combined immunodeficiency associated with alopecia in southern Italy population[J]. Ann Hum Genet,2004,68:265-268.
[3]Auricchio L,Adriani M,Frank J,et al. Nail dystrophy associated with a heterozygous mutation of the nude/SCID human FOXN1 (WHN) gene[J]. Arch Dermatol,2005,141:647-648.
[4]Du Q,Huynh LK,Coskun F,et al. FOXN1 compound heterozygous mutations cause selective thymic hypoplasia in humans[J]. J Clin Invest,2019,129:4724-4738.
[5]Giardino G,Sharapova SO,Ciznar P,et al. Expanding the Nude SCID/CID Phenotype Associated with FOXN1 Homozygous,Compound Heterozygous,or Heterozygous Muta-tions[J]. J Clin Immunol,2021,41:756-768.
[6]Flanagan SP. ‘Nude’,a new hairless gene with pleiotropic effects in the mouse[J]. Genet Res,1966,8:295-309.
[7]Nehls M,Pfeifer D,Schorpp M,et al. New member of the winged-helix protein family disrupted in mouse and rat nude mutations[J]. Nature,1994,372:103-107.
[8]Pignata C,Fiore M,Guzzetta V,et al. Congenital Alopecia and nail dystrophy associated with severe functional T-cell immunodeficiency in two sibs[J]. Am J Med Genet,1996,65:167-170.
[9]Markert ML,Marques JG,Neven B,et al. First use of thymus transplantation therapy for FOXN1 deficiency (nude/SCID): a report of 2 cases[J]. Blood,2011,117:688-696.
[10]Firtina S,Cipe F,Ng YY,et al. A Novel FOXN1 Variant Is Identified in Two Siblings with Nude Severe Combined Immunodeficiency[J]. J Clin Immunol,2019,39:144-147.
[11]Chou J,Massaad MJ,Wakim RH,et al. A novel mutation in FOXN1 resulting in SCID: a case report and literature review[J]. Clin Immunol,2014,155:30-32.
[12]Albuquerque AS,Marques JG,Silva SL,et al. Human FOXN1-deficiency is associated with alphabeta double-negative and FoxP3+ T-cell expansions that are distinctly modulated upon thymic transplantation[J]. PLoS One,2012,7:e37042.
[13]Radha RDA,Panday NN,Naushad SM. FOXN1 Italian founder mutation in Indian family: Implications in prenatal diagnosis[J]. Gene,2017,627:222-225.
[14]Albar R,Mahdi M,Alkeraithe F,et al. Epstein-Barr virus associated with high-grade B-cell lymphoma in nude severe combined immunodeficiency[J]. BMJ Case Rep,2019,12:e227715.
[15]Tangye SG,Al-Herz W,Bousfiha A,et al. Human Inborn Errors of Immunity: 2019 Update on the Classification from the International Union of Immunological Societies Expert Committee[J]. J Clin Immunol,2020,40:24-64.
[16]Schorpp M,Hofmann M,Dear TN,et al. Characterization of mouse and human nude genes[J]. Immunogenetics,1997,46:509-515.
[17]Schuddekopf K,Schorpp M,Boehm T. The whn transcrip-tion factor encoded by the nude locus contains an evolutionarily conserved and functionally indispensable activation domain[J]. Proc Natl Acad Sci U S A,1996,93:9661-9664.
[18]Žuklys S, Handel A, Zhanybekova S,et al. Foxn1 regulates key target genes essential for T cell development in postnatal thymic epithelial cells[J]. Nat Immunol,2016,17:1206.
[19]Vigliano I,Gorrese M,Fusco A,et al. FOXN1 mutation abrogates prenatal T-cell development in humans[J]. J Med Genet,2011,48:413-416.
[20]Vaidya HJ,Briones LA,Blackburn CC. FOXN1 in thymus organogenesis and development[J]. Eur J Immunol,2016,46:1826-1837.
[21]Nowell CS,Bredenkamp N,Tetelin S,et al. Foxn1 regulates lineage progression in cortical and medullary thymic epithelial cells but is dispensable for medullary sublineage divergence[J]. PLoS Genet,2011,7:e1002348.
[22]Chen L,Xiao S,Manley NR. Foxn1 is required to maintain the postnatal thymic microenvironment in a dosage-sensitive manner[J]. Blood,2009,113:567-574.
[23]Cheng L,Guo J,Sun L,et al. Postnatal tissue-specific disruption of transcription factor FoxN1 triggers acute thymic atrophy[J]. J Biol Chem,2010,285:5836-5847.
[24]Larsen BM,Cowan JE,Wang Y,et al. Identification of an Intronic Regulatory Element Necessary for Tissue-Specific Expression of Foxn1 in Thymic Epithelial Cells[J]. J Immunol,2019,203:686-695.
[25]Markert ML,Alexieff MJ,Li J,et al. Complete DiGeorge syndrome: development of rash,lymphadenopathy,and oligoclonal T cells in 5 cases[J]. J Allergy Clin Immunol,2004,113:734-741.
[26]Zampieri M,Ciccarone F,Calabrese R,et al. Reconfiguration of DNA methylation in aging[J]. Mech Ageing Dev,2015,151:60-70.
[27]Pignata C,Gaetaniello L,Masci AM,et al. Human equivalent of the mouse Nude/SCID phenotype: long-term evaluation of immunologic reconstitution after bone marrow transplantation[J]. Blood,2001,97:880-885.

