【衡道丨病例】普通型脊索瘤(Conventional chordoma)如何诊断?
时间:2024-06-06 21:01:05 热度:37.1℃ 作者:网络
患者病史
患者男性,56岁,视物模糊5年余,加重2月就诊。
影像检查
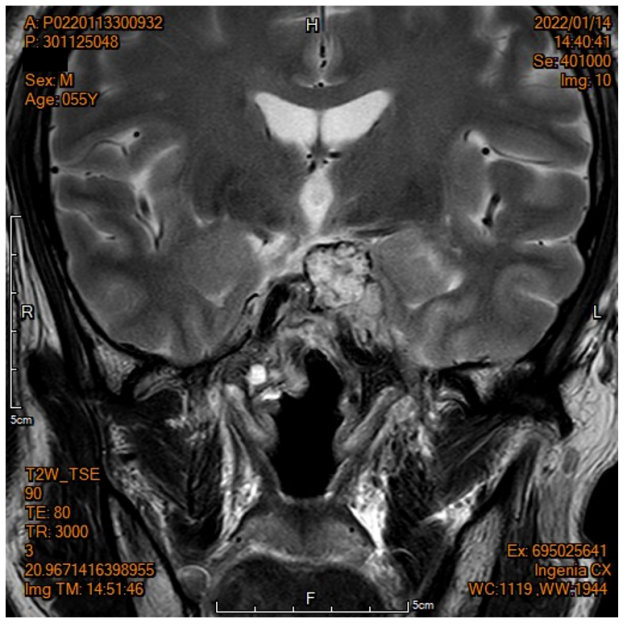
2021.11.11 鼻窦CT:
鞍区占位,累及视交叉、鞍上池、左侧海绵窦、桥前池等,鞍背骨质破坏,垂体瘤?
2021.11.12 鼻窦MRI:
鞍区左后上方软组织影,累及周围结构,部分包绕左侧颈内动脉海绵窦段,考虑低度恶性或良性可能。
大体形态
“鞍区肿瘤”:
灰白灰褐碎组织一堆,共大小1.0×1.0×0.2cm。
镜下形态

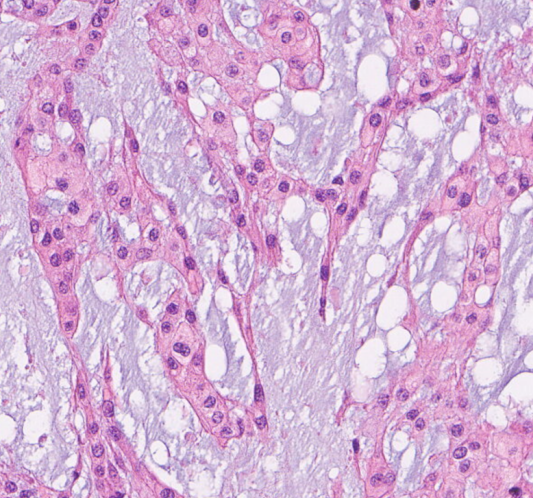
送检组织较碎,低倍镜下可见红染区及淡染区
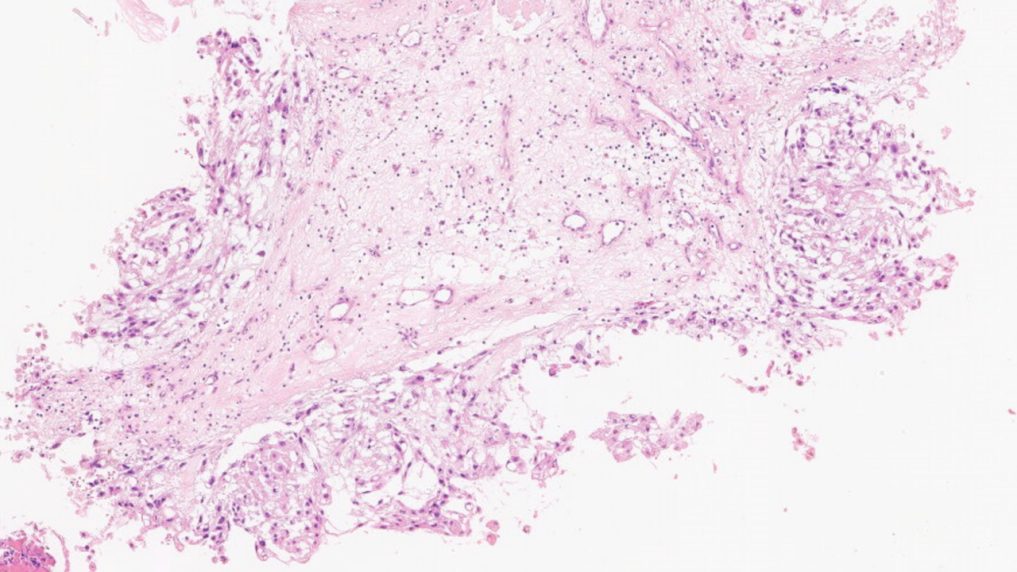
局部见肿瘤呈条索状排列
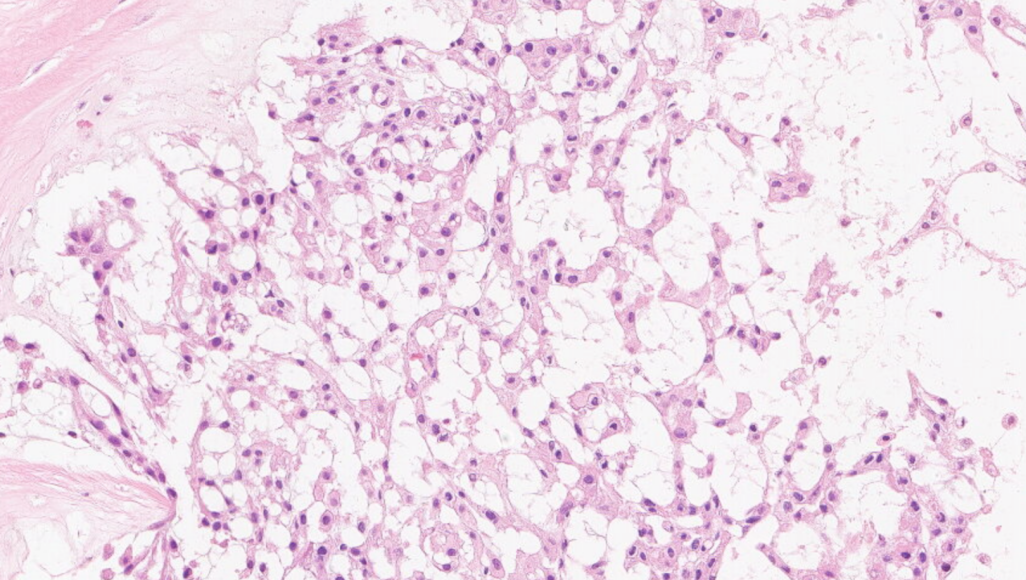
肿瘤细胞呈条索状排列,背景可见黏液样基质
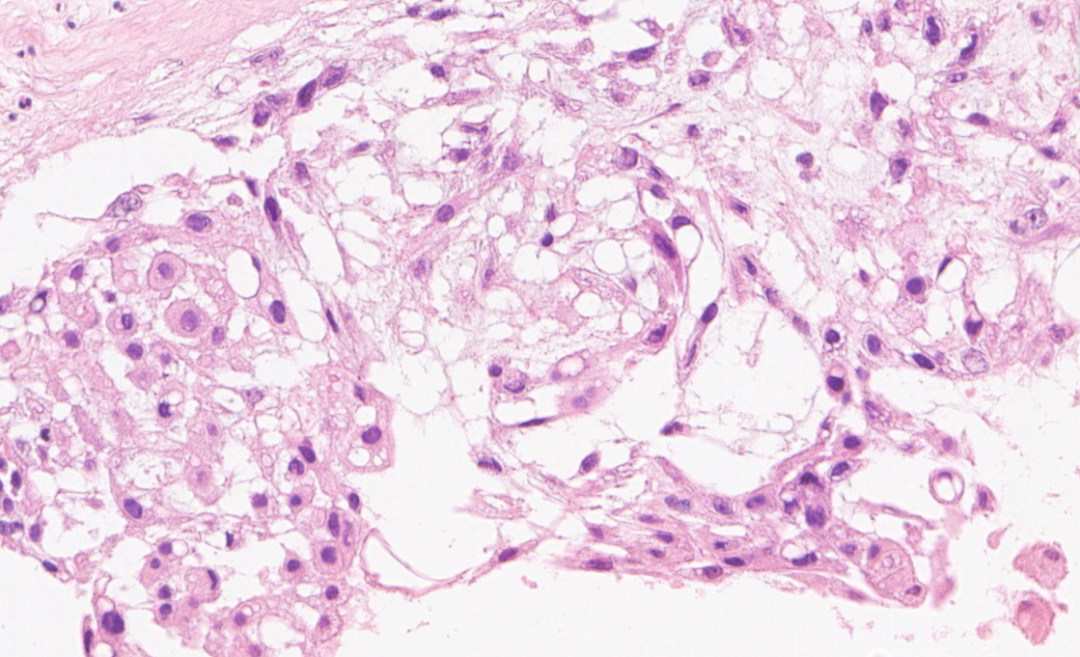
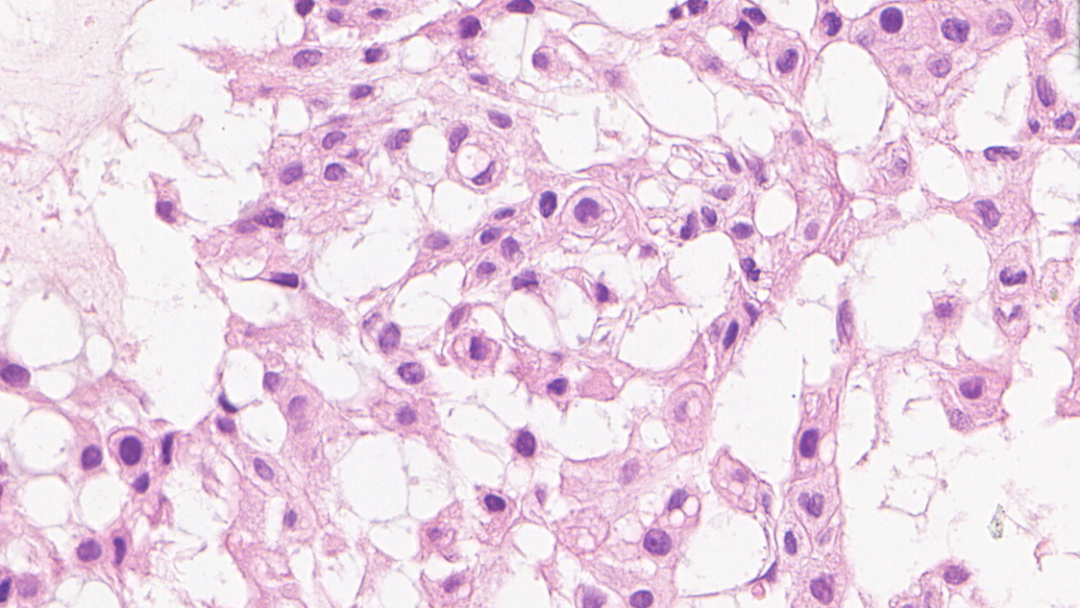
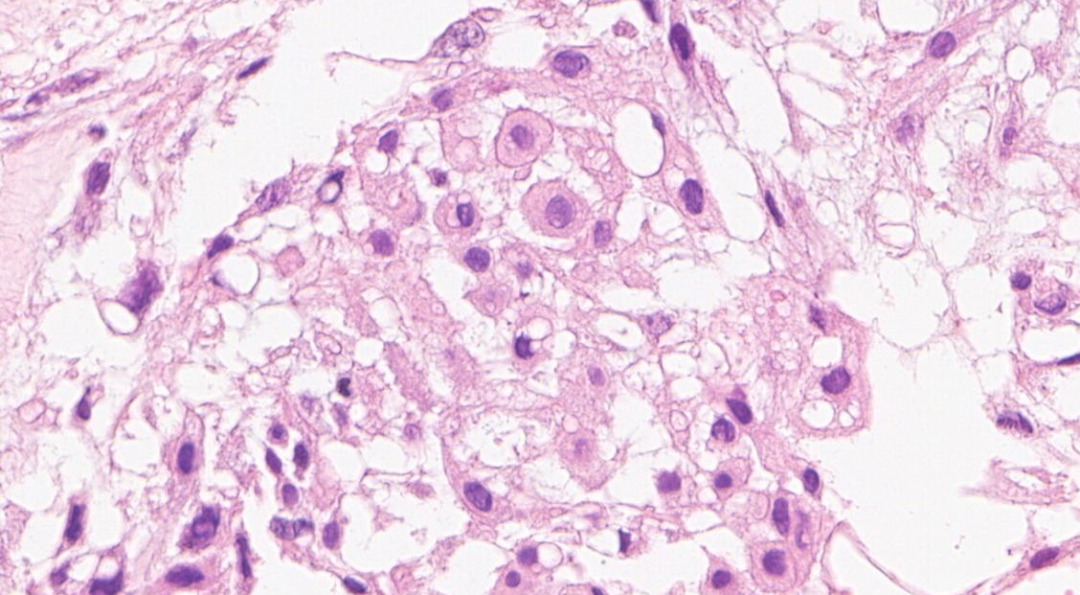
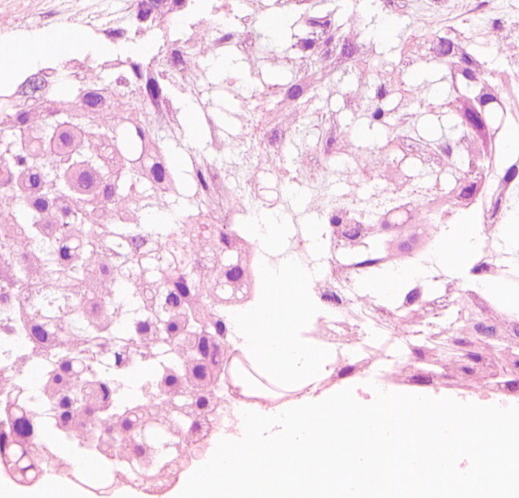
可见两类细胞:上皮样/星芒状细胞,空泡细胞
免疫组化
阳性标记 肿瘤细胞
1.AE1/AE3弥漫+
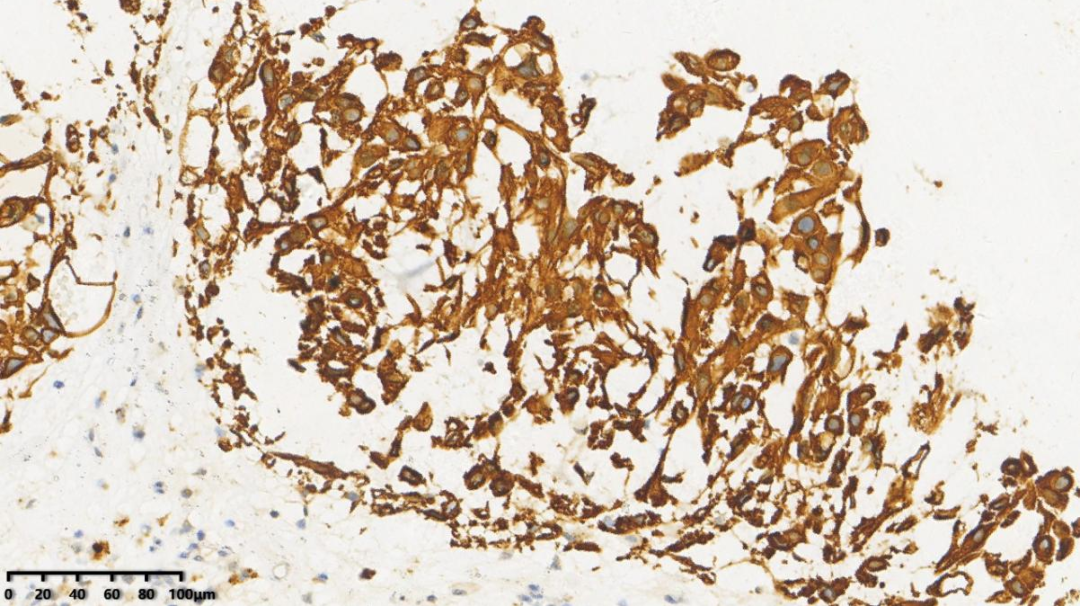
2.EMA弥漫+
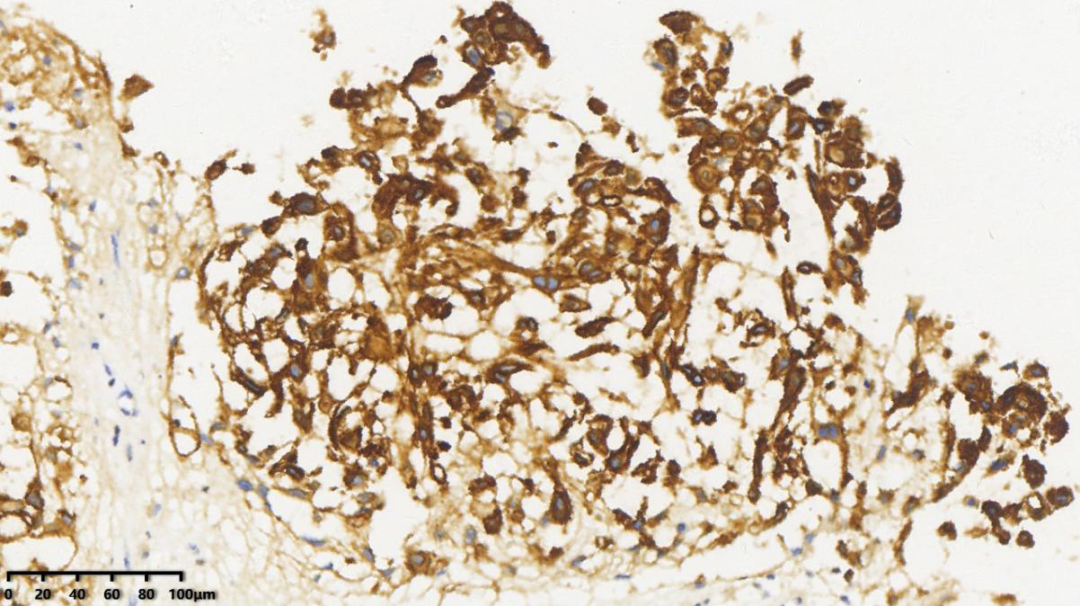
3.Vimentin+
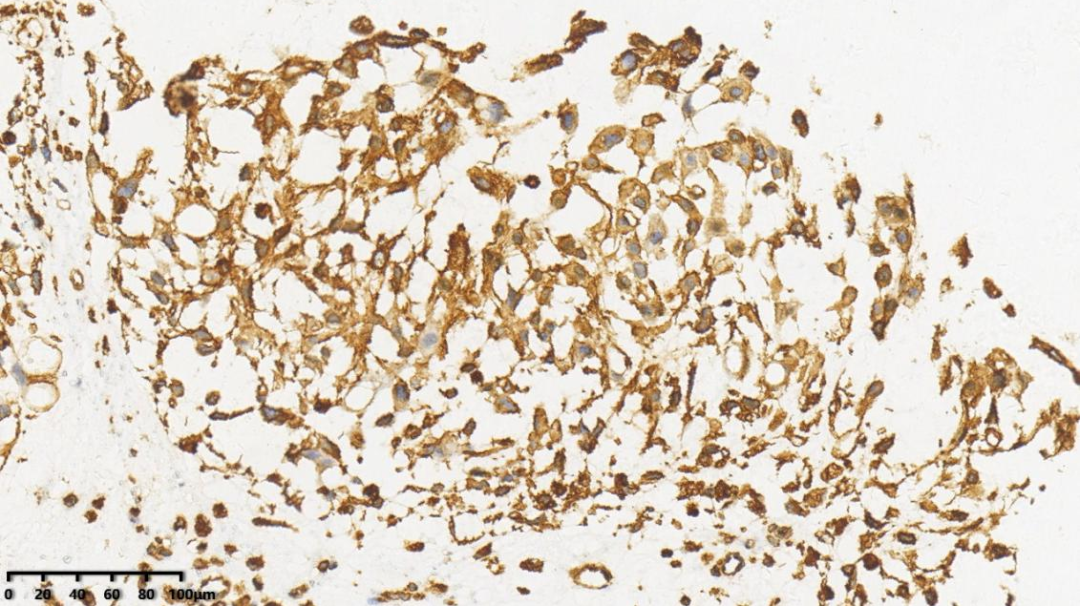
4.Brachyury细胞核+
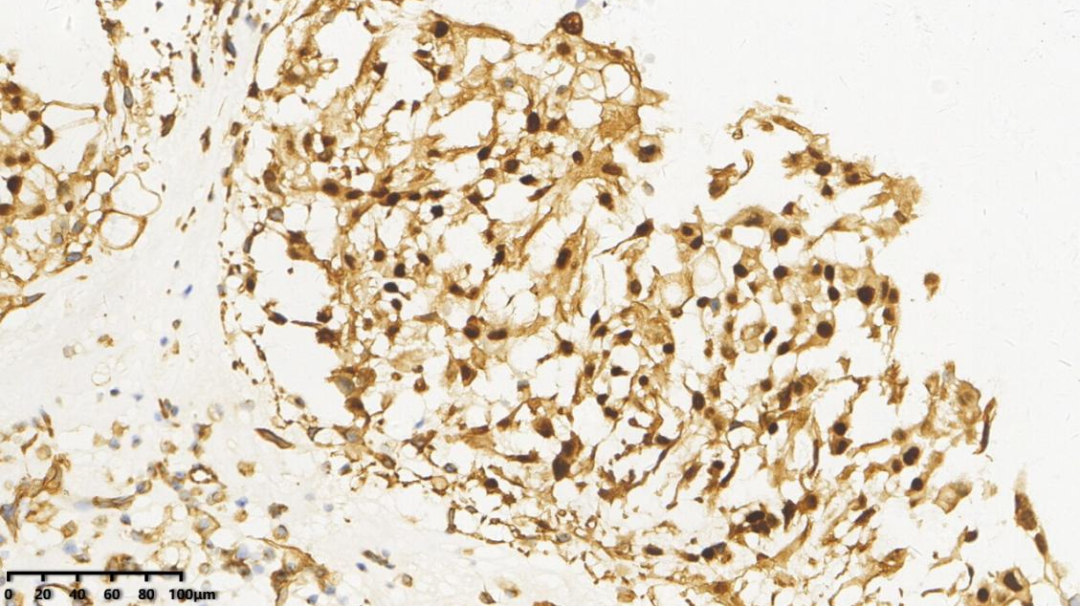
5.S-100少量
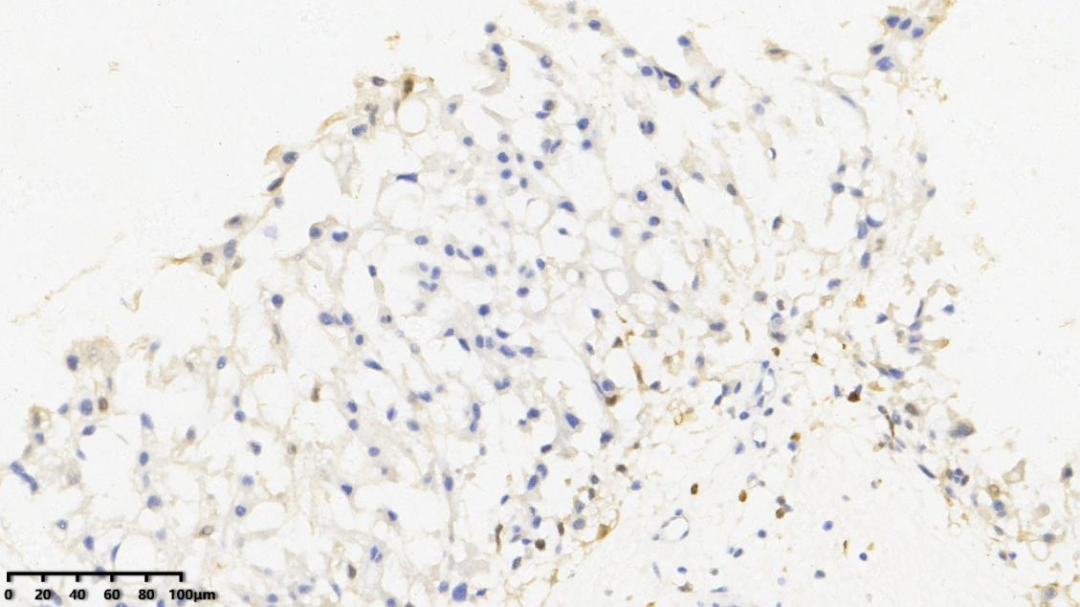
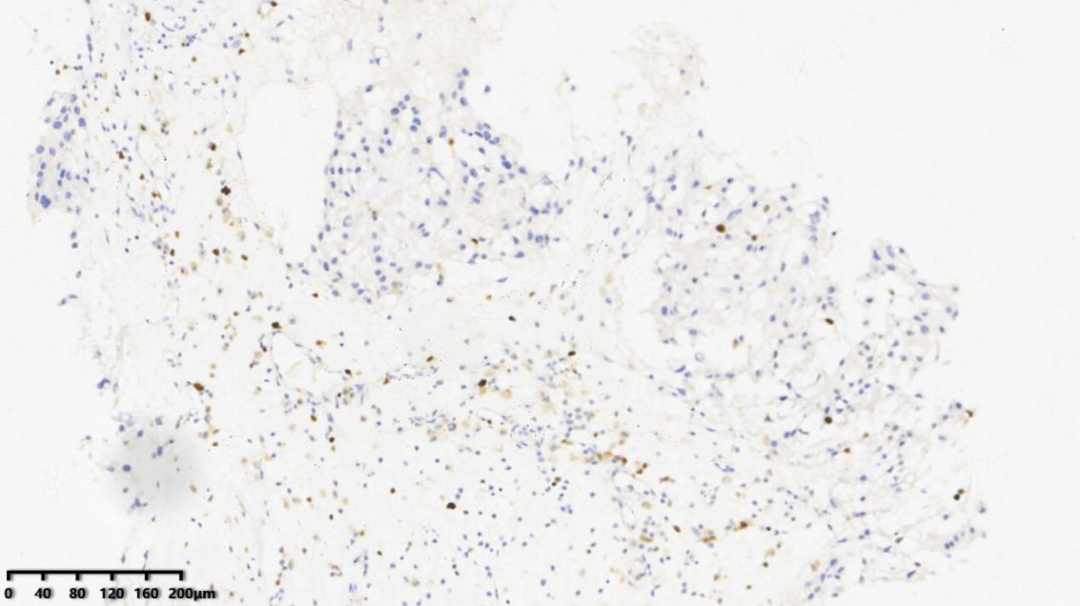
6.Ki67约1%
阴性标记
1.CgA -
2.SYN -
3.GFAP -
4.Olig-2 -
5.SMA -
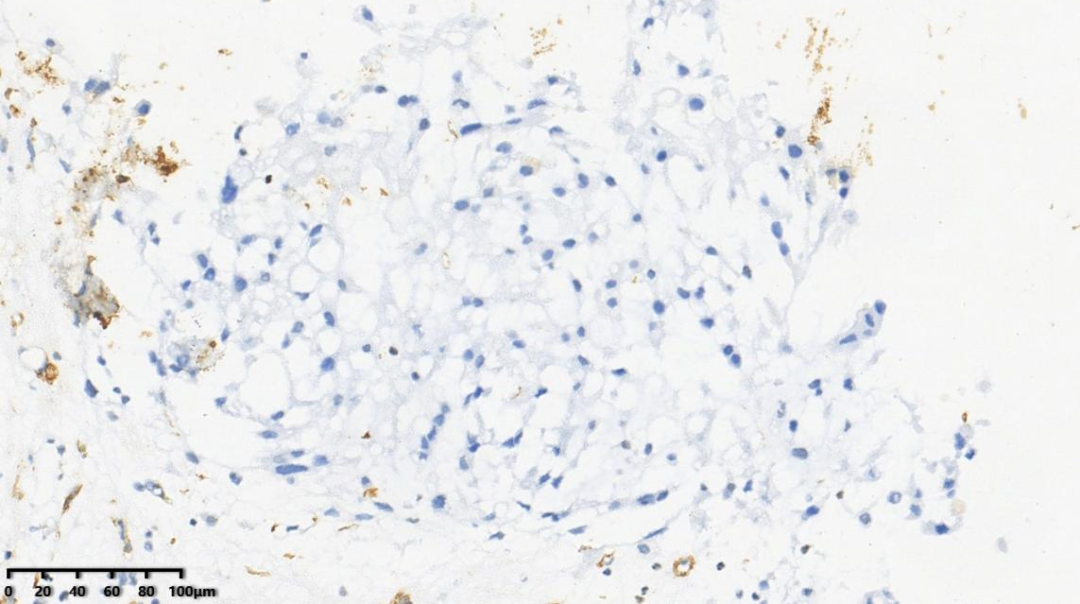
6.Desmin -
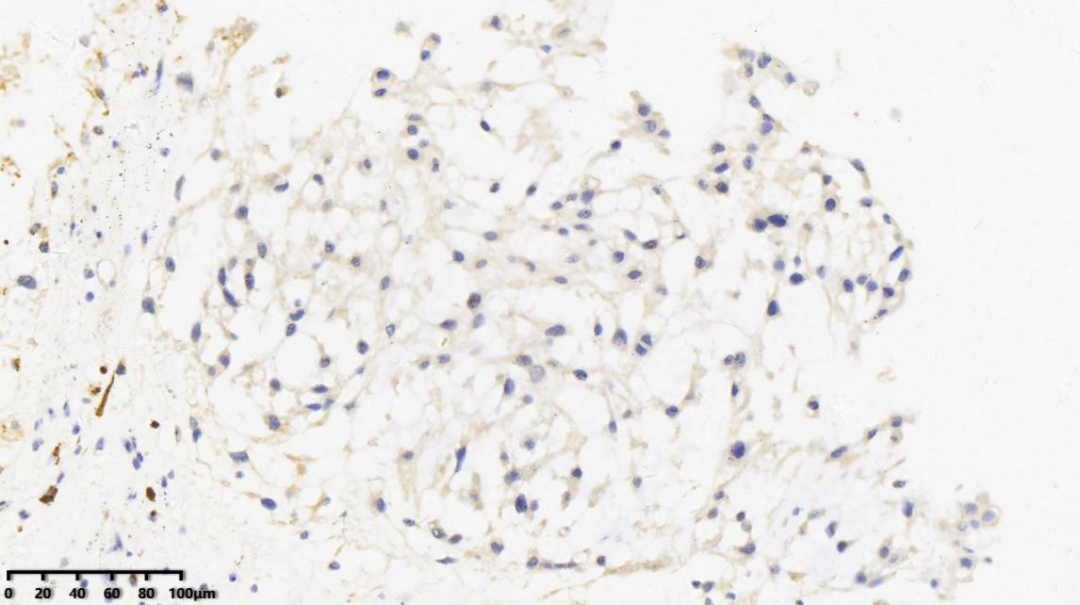
免疫组化结果汇总
阳性标记:
AE1/AE3、EMA、Vimentin、Brachyury、S-100(少量+)、Ki67(1%+)
阴性标记:
SMA、Desmin、CgA、SYN、GFAP、Olig-2
最终诊断
“鞍区肿瘤”:普通型脊索瘤。
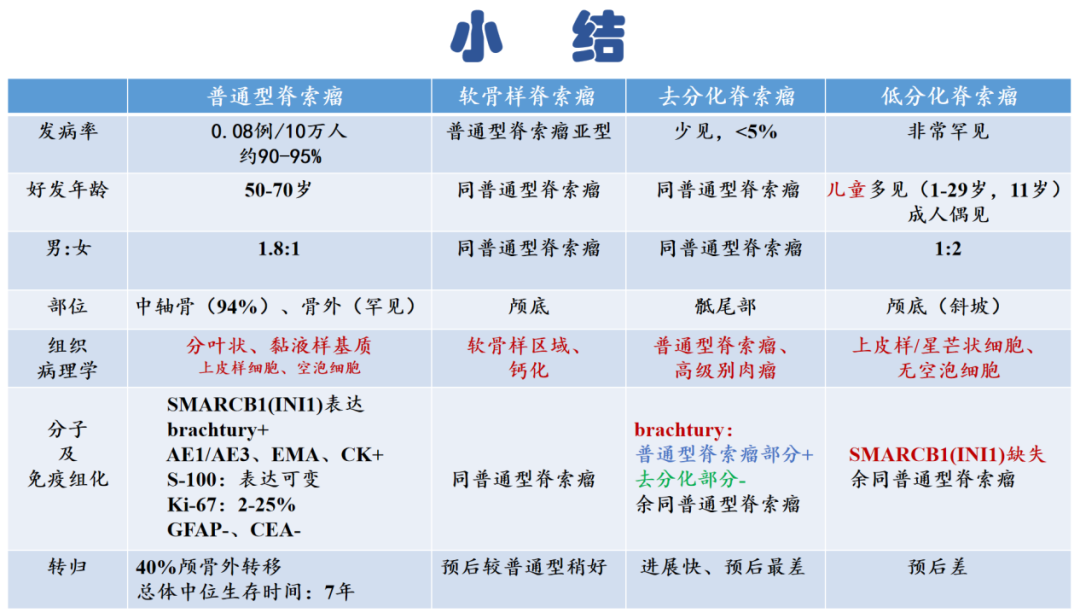
普通型脊索瘤诊断要点
定义
普通型脊索瘤(Conventional chordoma) ICD-O 9370/3
普通型脊索瘤是一种表现为脊索分化的恶性肿瘤,通常发生于中轴骨的骨骼中。
第五版WHO分类中将软骨样脊索瘤列为其亚型。
临床特点
好发年龄为50-70岁;发生部位主要位于融合节段脊柱,其中颅骨32%,脊柱32.8%,骶骨和尾骨29.2%,少数位于中轴骨外;儿童和青年人则主要发生于颅底、颈椎上部。
影像表现:
表现为分叶状、溶解性、破坏性病变,无钙化强回声;T1呈低信号(常包含高信号灶),T2呈高信号。
治疗方式:
手术(易复发),放疗(质子治疗),化疗(不敏感),药物治疗。
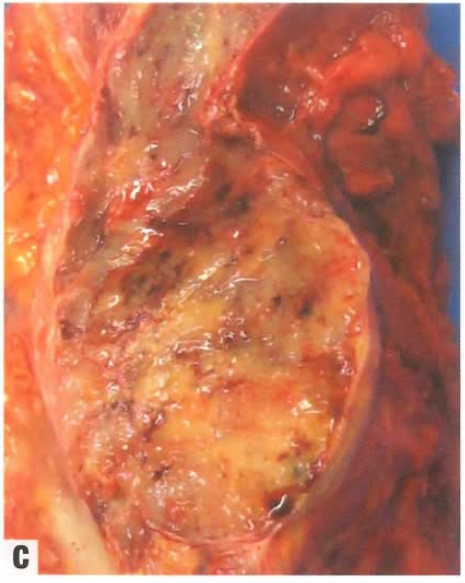
大体形态
分叶状肿块,胶冻样外观,侵及周围组织。
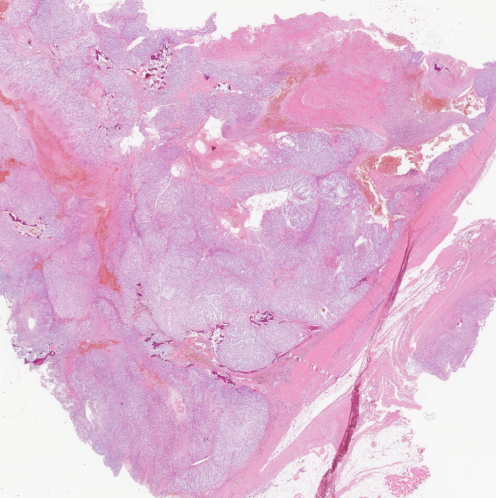
镜下形态
-
纤维分隔呈小叶状、结节状结构,黏液样细胞基质;
-
肿瘤细胞排列成条索状或带状;
-
两类细胞:上皮样/星芒状细胞,空泡细胞
免疫组化
阳性标记:AE1/AE3、EMA、CK、Vimentin、Brachyury(核+)、S-100(不定)、Ki67(2%-25%+);
阴性标记:SMA、Desmin、CgA、SYN、GFAP。
Brachyury介绍:
-
该蛋白定位于脊索来源的细胞;
-
在脊索组织、脊索良恶性肿瘤中均有近乎全面的表达(92%);
-
由TBXT(6q27)基因编码,是一种胚胎核转录因子,影响中胚层的形成和分化;
-
该基因的变异可引起神经管、脊柱发育缺陷,与脊索瘤的易感性有关;
-
其他阳性表达的恶性肿瘤:中枢神经系统血管母细胞瘤(胞浆+),睾丸生殖细胞肿瘤等。
分子改变
70%的病例在9p21处出现CDKN2A(p16, p14ARF)或CDKN2B(p15)纯合子或杂合子缺失;16%的病例中有PI3K信号通路激活;10%的病例中LKS失活。
诊断标准
基本标准:
与影像学一致;大量上皮样细胞表现出脊索分化,分布于黏液样基质中;表达brachyury和CK。
第五版WHO中关于脊索瘤的更新:

1、良性脊索细胞肿瘤
(Benign notochordal cell tumour)
(1)定义
一种显示脊索分化的良性肿瘤。
(2)临床特点
见于任何年龄,可能来自于未退化的脊索;
部位:颅底、脊椎、骶尾部;偶可发生在骨外(肺…);20%尸检可见。
(3)影像及大体形态
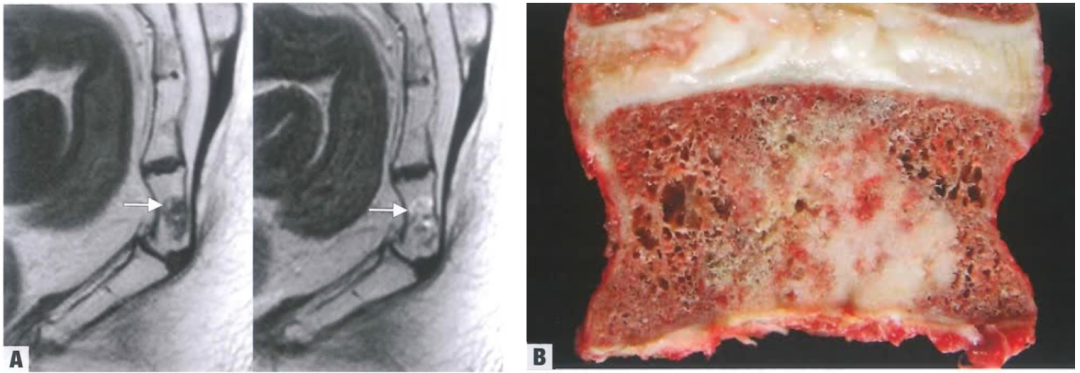
A:左侧T1加权与右侧T2加权像可见骨内病变,边界清楚,箭头所指高信号灶提示脂肪成分。B:肿瘤表现为白色致密、边界清楚的病灶。
(4)镜下形态
-
无分叶状结构、纤维间隔
-
无黏液样基质
-
无血管系统
-
无细胞异型
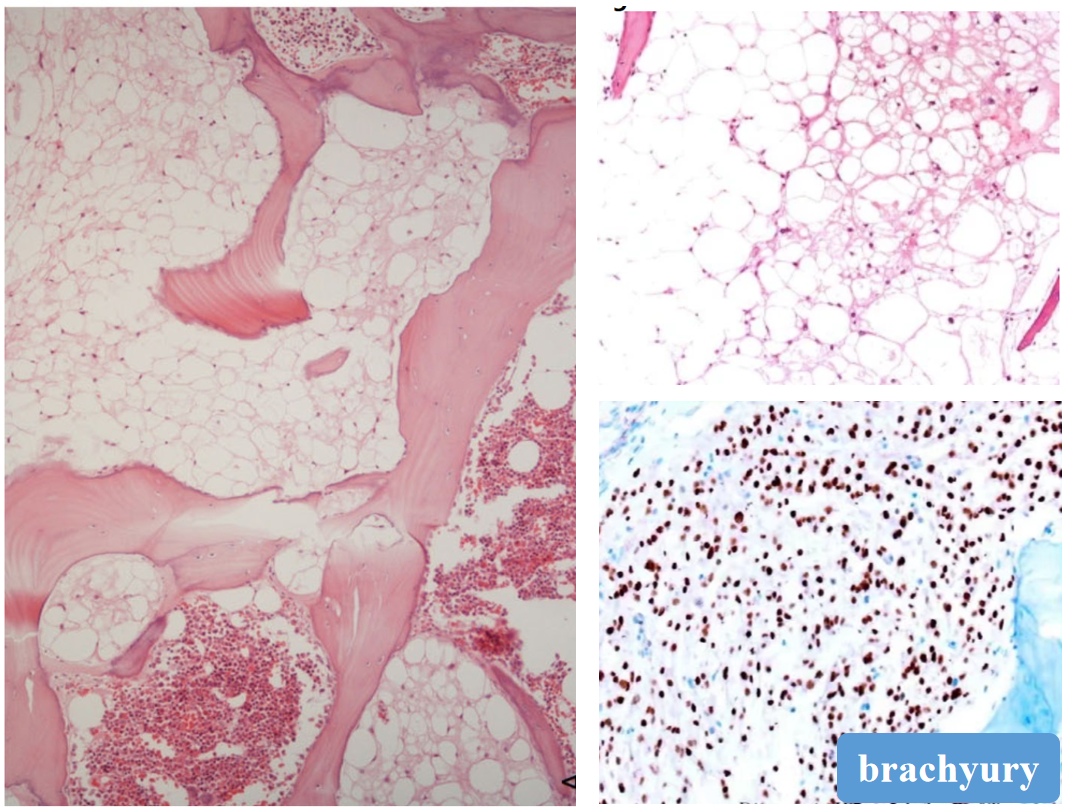
(5)免疫表型
表达AE1/AE3、S-100、EMA,特征性地表达brachyury。
(6)治疗及预后
预后良好,可不需手术治疗,长期随诊。
2、去分化脊索瘤
(Dedifferentiated chordoma)
(1)定义
普通型脊索瘤和高级别肉瘤同时存在的双向分化的脊索瘤。(肉瘤多为高级别梭形、多形性肉瘤,可出现骨肉瘤和横纹肌肉瘤)
(2)影像及大体形态
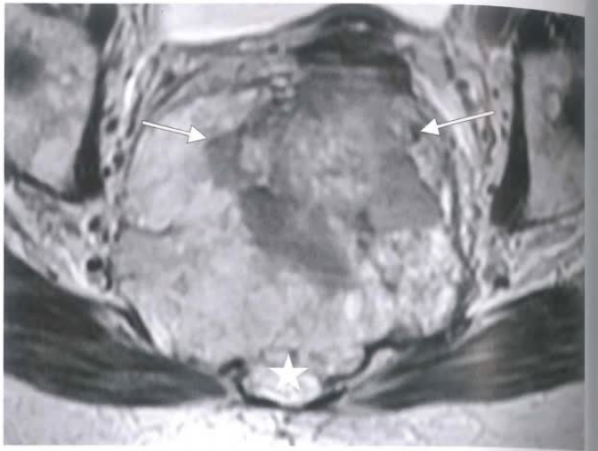
骨盆轴位T2加权像显示双向肿瘤。箭头所指肿瘤边界清晰,星号示肿块破坏骶前间隙。
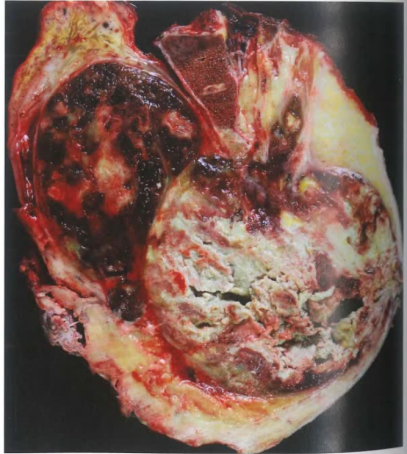
出血区为普通型脊索瘤区域,肉质区为去分化区域。
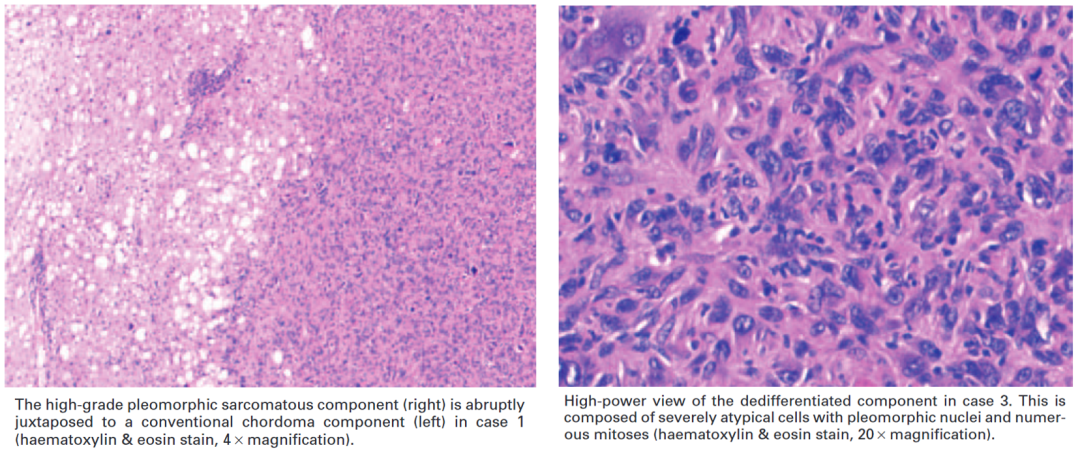
(3)镜下形态
(4)免疫表型
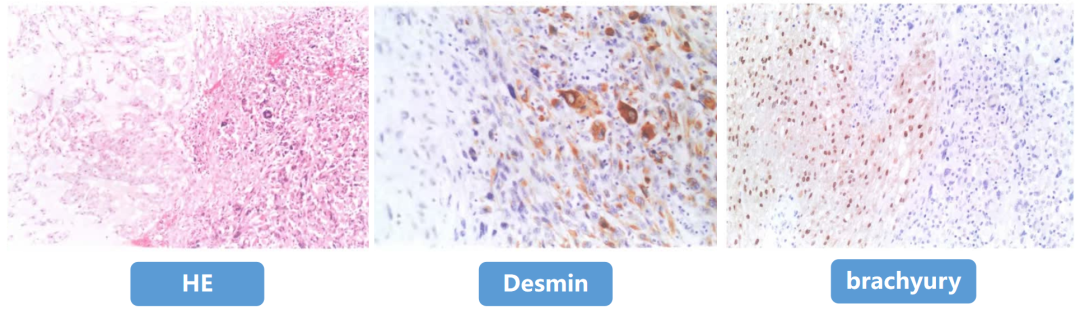
-
阳性:AE1/AE3、EMA、Vimentin、S-100(不定)、Ki67(2%-25%+);
-
阴性:SMA、Desmin、CgA、SYN、GFAP。
-
去分化成分:局灶表达上皮标记,但不表达brachyury。
3、低分化脊索瘤
(Poorly differentiated chordoma)
(1)定义
具有脊索分化的分化差的肿瘤,通常发生于中轴骨,以SMARCB1(INI1)表达缺失为特征。
(2)分子遗传学
大多数病例存在SMARCB1(INI1)缺失;部分病例有EWSR1位点共缺失;少数病例SMARCB1(INI1)完整,但缺乏表达,机制不清。
(3)临床特点
好发年龄:1-29岁,中位年龄11岁;
好发部位:主要位于颅底(斜坡)。
(4)影像学表现
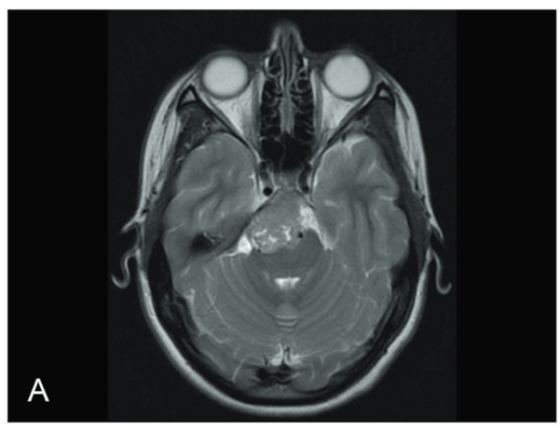
T2加权像显示颅底出现一个大的强化肿瘤。
(5)镜下形态
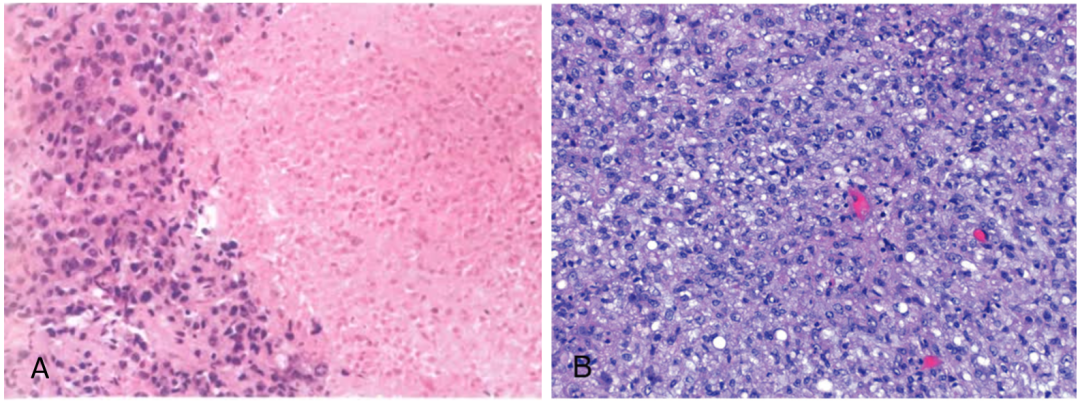
A:肿瘤由低分化的上皮样细胞组成,胞质嗜酸性,细胞核不规则,中央有坏死区,但无黏液样基质。B肿瘤细胞排列密集,核仁突出。
(6)免疫表型
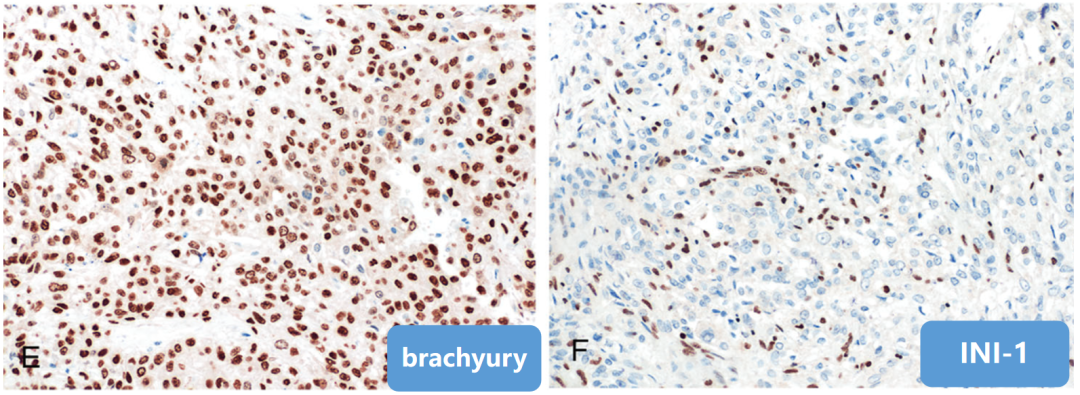
-
阳性:AE1/AE3、EMA、CK、Vimentin、Brachyury(核+)、S-100(不定)、Ki67(2%-25%+);
-
阴性:SMA、Desmin、CgA、SYN、GFAP;INI-1(缺失)。
鉴别诊断
在不同部位要想到鉴别不同的疾病,如下图所示:

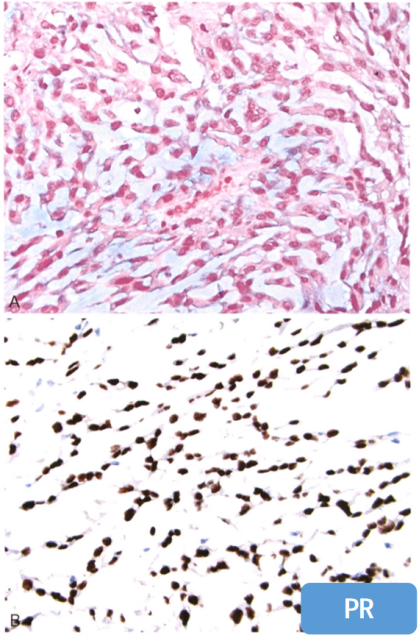
1.脊索样脑膜瘤
-
常混合经典型脑膜瘤;
-
呈条索状、小梁状结构;嗜碱性黏液间质;
-
多角形、上皮样等形态,部分呈空泡状;
-
免疫组化标记:
PR(+)、Vimentin(+)、EMA(+)、S-100(局灶+);
brachyury(-)、GFAP(-)、CD34(-)、OCT4(-)。
2.脊索样胶质瘤
-
巢状、线性实性排列;
-
黏液基质内淋巴细胞、浆细胞浸润;
-
免疫组化标记:
GFAP、TTF1(核+)、CD34、PAS阳性,
brachyury(-)。
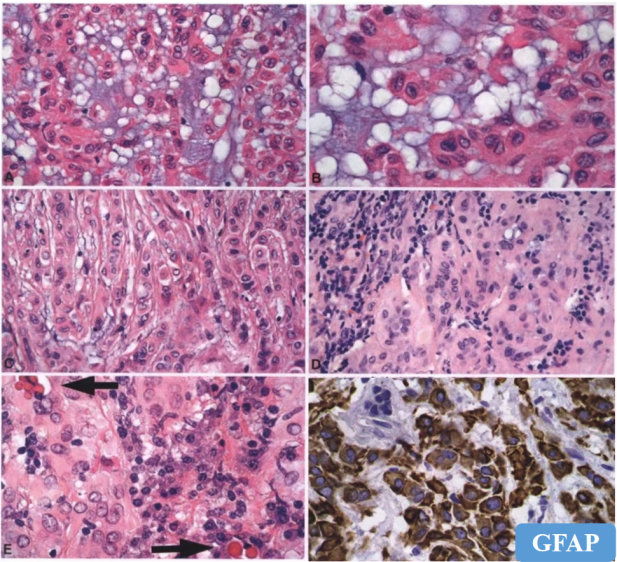
3.血管母细胞瘤
-
丰富小血管、大空泡间质细胞;
-
特殊染色:网状纤维、油红O脂肪染色;
-
免疫组化标记:
brachyury(胞浆+)、inhibin、D2-40、S-100、NSE阳性;
-
家族性血管母细胞瘤VHL双等位基因失活。
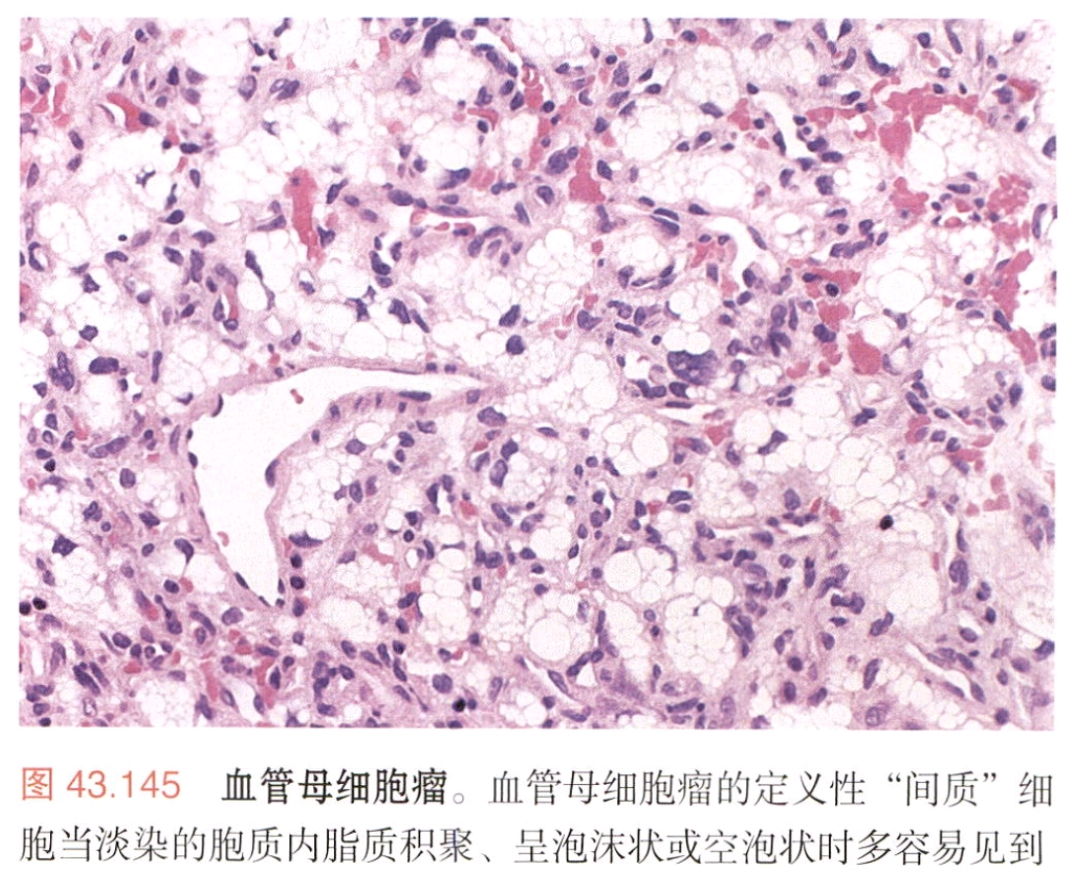
4.软骨肉瘤
-
Ⅰ级:细胞数量少、核小,软骨基质;
-
Ⅱ级:黏液样变性;
-
免疫组化标记:
SOX9(+)、SITB2 (+),
brachyury(-)、AE1/AE3(-)、EMA(-);
-
分子改变:IDH1或IDH2突变。
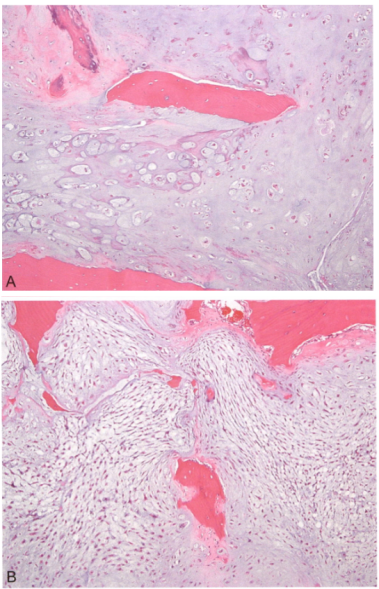
5.骨外黏液样软骨肉瘤
-
呈分叶状,胞浆嗜酸可有空泡;
-
大量黏液软骨样基质;
-
免疫组化标记:
CD117、SYN、NSE、PGP9.5阳性;
brachyury(-);
-
分子改变:NR4A3 重排。
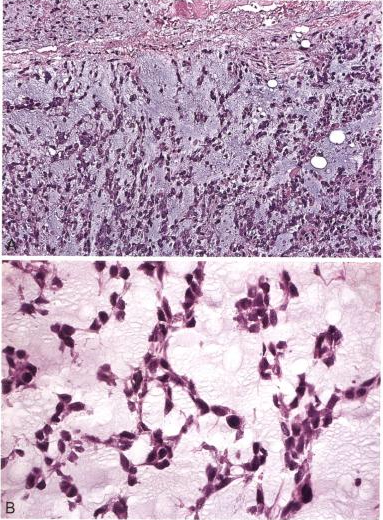
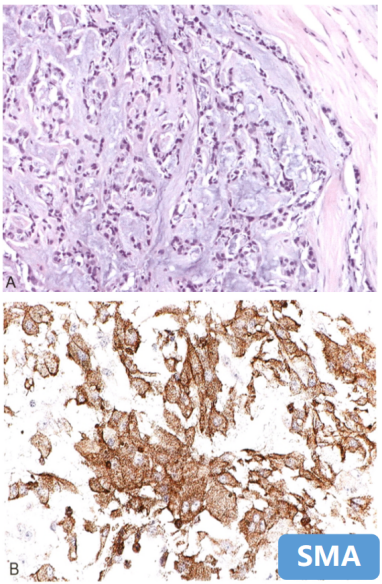
6、肌上皮瘤
-
呈结节状,肌上皮成分排列呈条索状,软骨或骨化生;
-
免疫组化标记:
Calponin、SMA、EMA、 GFAP、 p63阳性,
brachyury(-);
-
分子改变:可显示EWSR1 重排。
参考文献:
WHO5th Classification of Soft Tissue and Bone Tumours
ROSAI AND ACKERMAN’S SURGICAL PATHOLOGY 11th
DuJ, et al. J Clin Pathol 2018;0:1–9.
THE JOURNAL OF BONE AND JOINT SURGERY (2008) 90-B:652–656
Clinicopathologic characteristics of poorly differentiated chordoma[J]. Mod Pathol, 2018

