Translational Psychiatry:基于机器学习和深度学习方法对抑郁症相关血液DNA甲基化的全面多队列探索
时间:2024-07-28 23:00:42 热度:37.1℃ 作者:网络
抑郁症是一种复杂的精神疾病,受多种因素的影响,包括生活经历、人际关系以及生物学决定因素如遗传学和表观遗传学。目前,抑郁症的诊断主要依赖于精神病学评估,临床上尚无实验室生物标志物用于验证。这种缺乏可靠的实验室诊断方法使得区分重度抑郁症(MDD)患者与暂时性低落情绪或其他类似精神疾病患者变得困难。

本研究利用八个不同的队列(包括六个公共队列和两个国内队列,共1942个样本)进行血液DNA甲基化的mega分析和meta分析,以评估抑郁症的甲基化标志物。通过使用12种机器学习和深度学习方法,对数据进行跨验证(CV)和持出测试,构建了包含有偏和无偏特征选择的模型。研究中采用的机器学习方法包括随机森林、支持向量机(SVM)和深度神经网络(DNN),而深度学习方法则包括多层感知器(MLP)和自编码器。
数据处理分为两个阶段:首先是数据的预处理,包括批次效应的校正和数据标准化;其次是特征选择,主要采用limma作为特征选择策略。此外,还探讨了不同特征选择方法对模型性能的影响。所有模型的训练和优化过程均在不同的交叉验证折中进行,以评估其泛化性能。
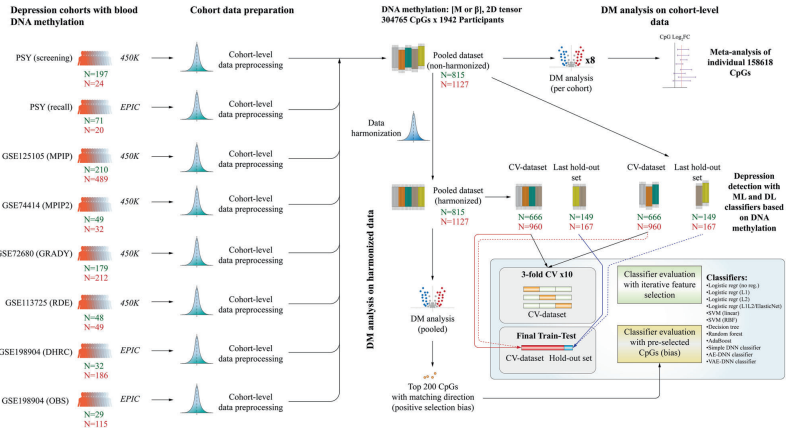
图1.本研究的工作流程概述
在mega分析和meta分析中,发现了1987个与抑郁症相关的CpG位点,这些位点在基因富集分析和eQTM数据中与轴突导向和免疫通路相关。随机森林分类器在批次处理的数据中表现最佳,在CV和持出测试中分别达到了0.73和0.76的AUC。然而,当使用标准化数据时,所有分类器在CV中的AUC均低于0.57,在持出测试中没有预测能力。
在特征预选的模型中,所有模型的性能显著提升(AUC增加超过14%),某些模型(如联合自编码器分类器)在最终测试中AUC达到了0.91,不受数据处理方法的影响。不同算法的特征选择方法(如L1惩罚逻辑回归)在某些情况下也优于limma。结果表明,随机森林模型在所有数据处理策略中均表现出色,适用于DNA甲基化评分。
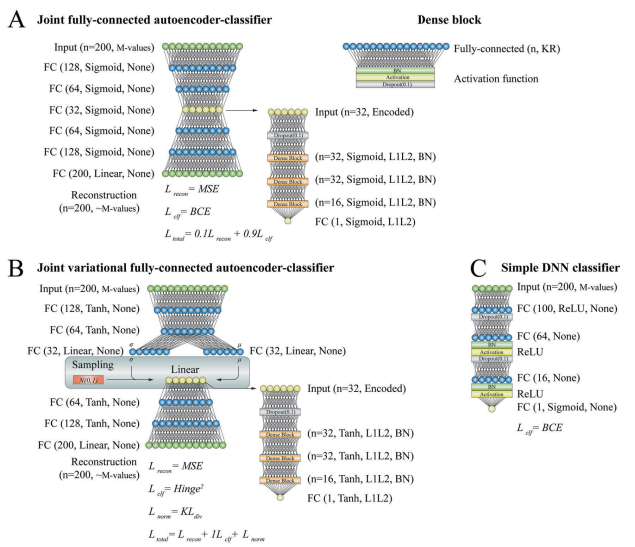
图2.深度学习模型的配置
本研究概述了未来可能用于抑郁症的生物标志物,并强调了DNA甲基化抑郁症分析中的许多重要方法学问题,包括机器学习策略的使用。研究结果显示,随机森林分类器在不同数据处理策略下均表现良好,而复杂的深度学习模型在小数据集上的表现不如树模型。这些发现为未来的抑郁症生物标志物研究提供了有价值的见解,并指出了数据处理和特征选择对模型性能的关键影响。
原始出处:
A.V. Sokolov and H.B. Schiöth. Decoding depression: a comprehensive multi-cohort exploration of blood DNA methylation using machine learning and deep learning approaches. Translational Psychiatry, 2024, 14:287. https://doi.org/10.1038/s41398-024-02992-y.

