Cell子刊:糖尿病治疗新突破!科学家发现了独立于胰岛素之外的新降糖途径
时间:2022-01-10 17:15:10 热度:37.1℃ 作者:网络
100年前胰岛素的发现为数百万糖尿病患者打开了一扇通往生命和希望的大门。从那时起,由胰腺产生的胰岛素一直被认为是治疗以高血糖(葡萄糖)为特征的疾病(例如糖尿病)的主要手段。
令人兴奋的是,近日来自美国的索尔克生物研究所、荷兰的格罗宁根大学等研究机构的专家惊奇地发现了第二种降糖分子,它产生于脂肪组织中,与胰岛素一样,也能有效而快速地调节血糖。他们的发现可能导致治疗糖尿病的新疗法的开发,也为代谢研究中有前途的新途径奠定了基础。
该研究发表在国际知名期刊Cell Metabolism上,研究表明一种名为FGF1的激素通过抑制脂肪分解来调节血糖。和胰岛素一样,FGF1通过抑制脂肪分解来控制血糖,但这两种激素的作用方式不同。更重要的是,这种独特的差异可以使FGF1能够安全和成功地降低胰岛素抵抗患者的血糖。

研究成果 (图源:Cell Metabolism)
在此之前,该团队研究人员发现外周给药FGF1可通过脂肪FGF受体1的介导迅速降低糖尿病小鼠模型的血糖水平,但其作用机制尚不清楚。在这篇文章中,他们进一步探究了这些现象背后的机制以及它们之间的联系。
首先,研究人员为了验证FGF1 诱导的葡萄糖降低依赖于脂肪组织中FGFR1的表达,FGFR1在成熟脂肪细胞中被选择性敲除(adR1KO小鼠)。研究结果显示,FGF1迅速降低饮食诱导肥胖(DIO) 野生型(WT)小鼠的血糖水平,但未能影响adR1KO小鼠的血糖,这进一步验证了他们之前的研究结果。
鉴于adR1KO小鼠中胰岛素水平的增加,以及肝葡萄糖生成(HGP)和脂肪分解之间的联系,研究人员假设FGF1可能影响脂肪分解。为了探索这一概念,研究人员起初要确定是否FGF1基因敲除(F1KO)小鼠的脂解作用受到干扰。体外脂肪分解实验显示,F1KO小鼠的性腺脂肪组织(gWAT)的脂肪分解显著增加。
此外,FGF1显著抑制基础和异丙肾上腺素(ISO)诱导的小鼠和源于脂肪细胞的人基质血管成分(SVF)的脂解反应,与脂肪细胞的内在效应一致。类似地,FGF1 以剂量依赖性方式抑制3T3-L1脂肪细胞中ISO诱导的脂解作用,这种作用被FGFR1抑制阻断。
为了确定外源性FGF1是否可以类似地影响体内脂肪分解,研究人员将DIO adR1WT和adR1KO小鼠在注射FGF1之前禁食一夜,以尽量减少胰岛素的代偿性变化。研究结果显示,FGF1将adR1WT小鼠的血清游离脂肪酸水平降低了约 30%,但未能影响adR1KO小鼠。此外,FGF1以脂肪FGFR1依赖性方式抑制体外脂肪分解。作为体内脂肪分解的衡量标准,研究人员将FGF1预处理的adR1WT和 adR1KO小鼠都注射了放射性标记的油酸。在 FGF1处理的adR1WT小鼠中,油酸的部分转换率降低,表明基础脂肪分解较低。相比之下,adR1KO小鼠的油酸转换不受 FGF1预处理的影响。上述研究数据表明,FGF1-FGFR1信号传导是一种调节脂肪分解的新途径。
为了确定FGF1对脂解作用的抑制是否会显著降低HGP,研究人员探究了FGF1影响糖异生的能力。研究结果显示,用FGF1预处理的ob / ob小鼠从丙酮酸合成葡萄糖的能力显著降低(丙酮酸耐受性测试,PTT),而当甘油是外源性底物时(甘油耐受性测试,甘油 TT),则没有发现差异。此外,FGF1抑制丙酮酸利用的能力取决于脂肪细胞FGFR1 的表达。
鉴于以上对丙酮酸和甘油利用的不同差异,使研究人员将目光聚焦到丙酮酸的下游。研究人员通过质谱法测量ob / ob小鼠肝脏中的糖异生中间体水平。丙酮酸下游的中间体,包括葡萄糖 6-磷酸(G6P)、果糖 6-磷酸 (F6P)、磷酸甘油酸 (PG)、磷酸烯醇丙酮酸(PEP)和草酰乙酸(OAA)在注射FGF1的小鼠中减少,而参与三羧酸的代谢物酸循环(TCA) 循环不受影响。此外,丙酮酸羧化酶变构激活剂乙酰辅酶A的水平降低。这些糖异生底物的减少主要在 adR1WT小鼠中重现,而adR1KO小鼠对FGF1的处理不敏感。另外,乙酰辅酶A的脂肪FGFR1依赖性降低伴随着丙酮酸羧化酶活性降低约50%。
研究人员为了探究这些发现与葡萄糖稳态的相关性,在短期连续 FGF1 给药(0.5毫克/千克,每隔一天,一周)后,对ob/ob小鼠进行了高胰岛素钳夹实验。研究结果显示,内源性葡萄糖产生 (EGP) 减少了约 25%。在钳夹条件下,用FGF1处理的小鼠需要更高的外源性葡萄糖输注率(GIR)来维持葡萄糖设定点,这种影响主要归因于 EGP 的减少,因为葡萄糖代谢清除率(GDR) 未改变。上述研究数据表明,FGF1以脂肪FGFR1依赖性方式抑制HGP。
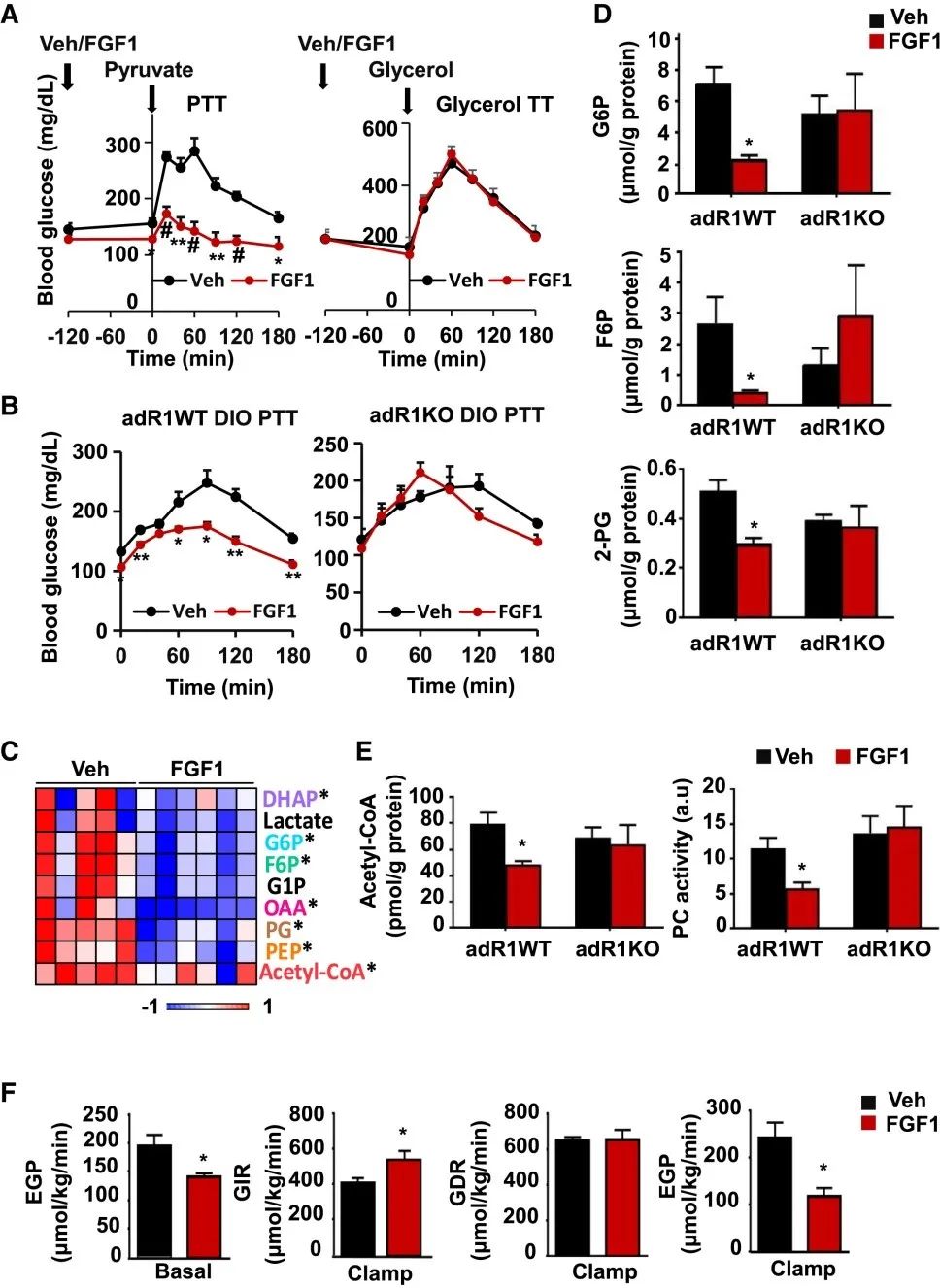
FGF1以脂肪FGFR1依赖性方式抑制HGP(图源:Cell Metabolism)
胰岛素通过PDE3B的PI3K依赖性激活抑制脂肪分解。由于FGFR1激活也可以通过PI3K途径发出信号,因此研究人员探究了FGF1的抗脂解作用是否受到PI3K抑制剂wortmannin的影响。研究结果显示,与胰岛素信号通路相平行,抑制剂wortmannin消除了FGF1诱导的3T3-L1脂肪细胞中FFA释放的减少。此外,在基于CRE荧光素酶的报告基因实验显示,FGF1减弱了ISO诱导的cAMP和 cAMP/PKA信号传导的增加,暗示可能对磷酸二酯酶活性产生影响。有趣的是,PDE3B的抑制并未损害FGF1诱导的脂解抑制。相比之下,FGF1的抗脂解活性被3T3-L1脂肪细胞以及小鼠和人类SVF衍生的脂肪细胞中的PDE4选择性抑制剂阻断。研究人员为了进一步确定FGF1诱导的体内脂解抑制是否需要PDE4活性,在FGF1注射前1小时,给DIO小鼠灌胃PDE4抑制剂。研究结果显示,PDE4抑制阻断FGF1的抑制脂肪分解的能力。
鉴于上述发现,研究人员推测FGF1-PDE4信号调节激素敏感性脂肪酶(HSL)磷酸化。研究结果显示,在基础细胞和ISO刺激的细胞中,FGF1抑制HSL 磷酸化的能力在PDE4抑制剂罗氟司特的存在下丧失了。
为了进一步监测FGF1在活细胞中影响pHSL-perilipin相互作用的能力,研究人员使用结合人脂联素启动子/增强子的腺相关病毒(AAV) 载体来表达GFP标记的 perilipin (perilipin-GFP)和3T3-L1脂肪细胞中带有mCherry标记的HSL(HSL-mCherry)。研究结果显示,FGF1减少了ISO诱导的perilipin-GFP和 HSL-mCherry的共定位,如荧光重叠的时间监测。在没有HSL融合的情况下,表达perilipin-GFP和mCherry的细胞中没有看到任何影响。此外,虽然选择性抑制 PDE4 或PDE3增加了周脂素-HSL共定位,与增加的 cAMP 水平和PKA激活一致,但只有PDE4抑制消除了FGF1效应。
基于之前将PDE4D与脂肪分解联系起来的研究,接下来研究人员探索了 PDE4D的过度表达是否足以概括FGF1抑制脂肪分解的能力。研究结果显示,3种PDE4D同种型的过表达以剂量依赖性方式抑制了3T3-L1脂肪细胞的脂解作用,其中 PDE4D3 同种型显示出最高的功效。为了继续加深对这一发现的理解,研究人员构建了限制PDE4D3在成熟脂肪细胞中表达的adAAV 载体。研究结果显示,adAAV-PDE4D3驱动的表达有力地抑制了ISO诱导的3T3-L1脂肪细胞中脂解、cAMP 和perilipin-GFP / HSL-mCherry共定位的增加。上述研究数据表明,FGF1 通过激活 PDE4 抑制 cAMP-PKA 通路。
FGF1诱导的脂解抑制依赖于PDE4的发现增加了这一途径有助于胰岛素抵抗小鼠的葡萄糖稳态的可能性。为了探索这一可能性,研究人员用PDE4抑制剂罗氟司特治疗随机喂养的DIO小鼠。研究结果显示,抑制PDE4会瞬时增加血糖、血清FFA和胰岛素水平。值得注意的是,罗氟司特预处理ad lib喂养的小鼠后,FGF1降低血糖水平的能力丧失了。相比之下,PDE3的抑制未能影响FGF1调节葡萄糖水平的能力。
鉴于PDE4D在体内调节脂解的能力,研究人员进一步探索了该PDE家族在FGF1代谢活动中的作用。研究结果显示,来自 PDE4DKO 小鼠的脂肪外植体显示出更高的基础和 ISO 刺激的脂解作用,而PDE4DKO SVF 来源的脂肪细胞对 FGF1处理不敏感。更重要的是,FGF1未能降低这些喂食 HFD 的PDE4DKO小鼠的血糖,而这一缺陷通过脂肪组织中由 adAAV 驱动的PDE4D3 表达得以恢复。上述研究数据表明,FGF1诱导的脂解和血糖抑制依赖于体内PDE4D。
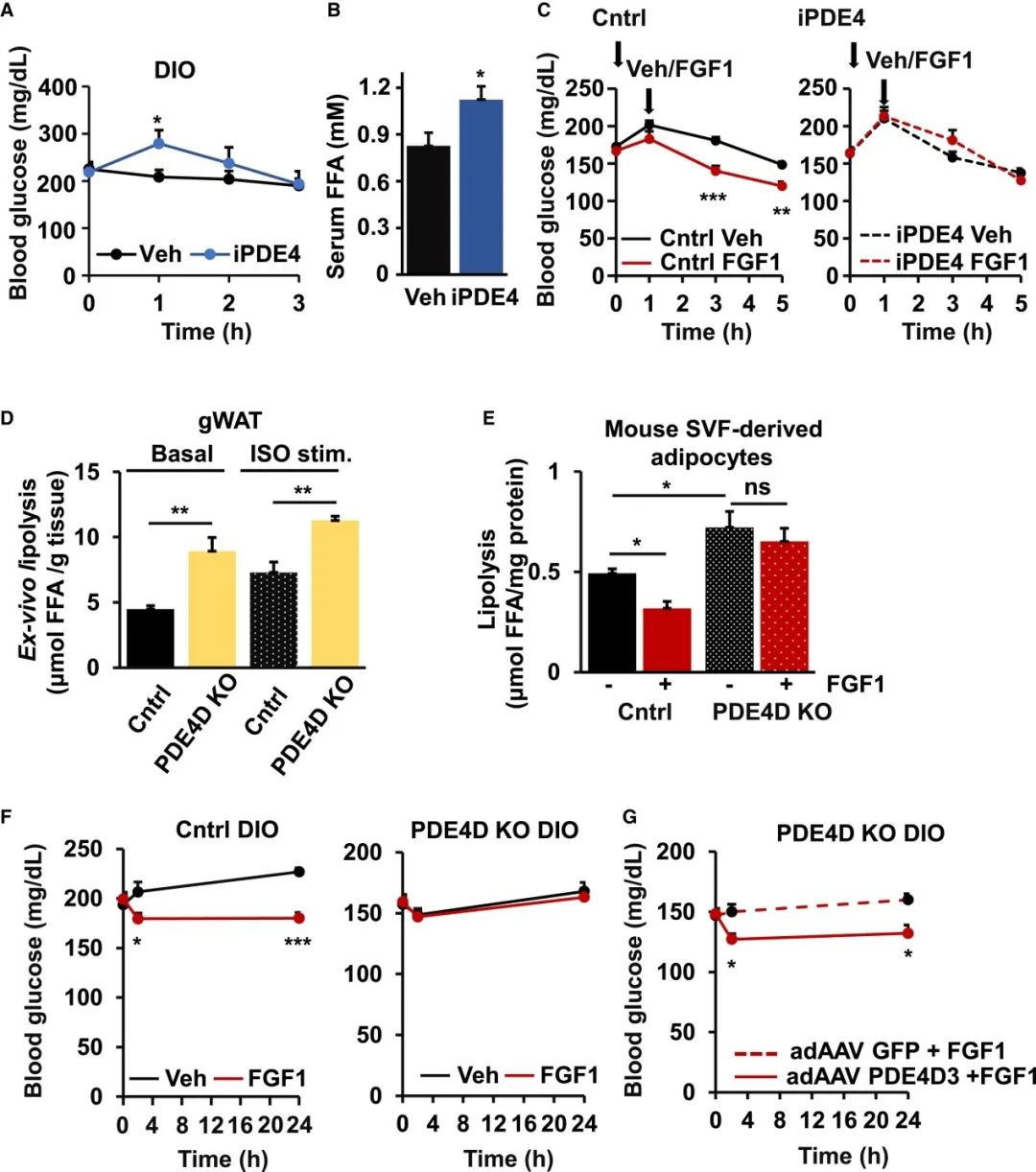
FGF1诱导的脂解和血糖抑制依赖于PDE4D(图源:Cell Metabolism)
接下来,研究人员又通过一系列实验证明了FGF1/PDE4D通路的抗脂解活性需要 PDE4D在S44处的特异性磷酸化,从而发挥作用。为了证实这一发现的体内相关性,研究人员向ob/ob小鼠注射了adAAV-GFP、adAAV-PDE4D3或 adAAV-PDE4D3 S44A。研究结果显示,PDE4D3的过表达导致了较低的即兴喂食和过夜禁食血糖和血清FFA水平,以及在进食状态向低胰岛素水平的趋势。相比之下,S44A 突变体的过度表达未能影响这些代谢参数。重要的是,FGF1 在过度表达 PDE4D3 的小鼠中诱导了更大的葡萄糖水平降低,而表达 S44A 突变体的小鼠的反应与对照小鼠的反应没有区别。
研究人员为了将内源性FGF1信号与S44磷酸化联系起来,在隔夜禁食和再喂养条件下从食物和HFD喂养的小鼠中收集gWAT库。研究结果显示,重新喂养的小鼠的pS44水平几乎翻了一倍。有趣的是,HFD显著降低了禁食和进食状态下的 S44 磷酸化,表明 PDE4D 在胰岛素抵抗高脂血症中的作用。上述研究数据支持外源性FGF1通过以PDE4D3依赖的方式抑制脂肪分解来降低血糖水平的机制,并且这种机制与摄食的生理反应有关。
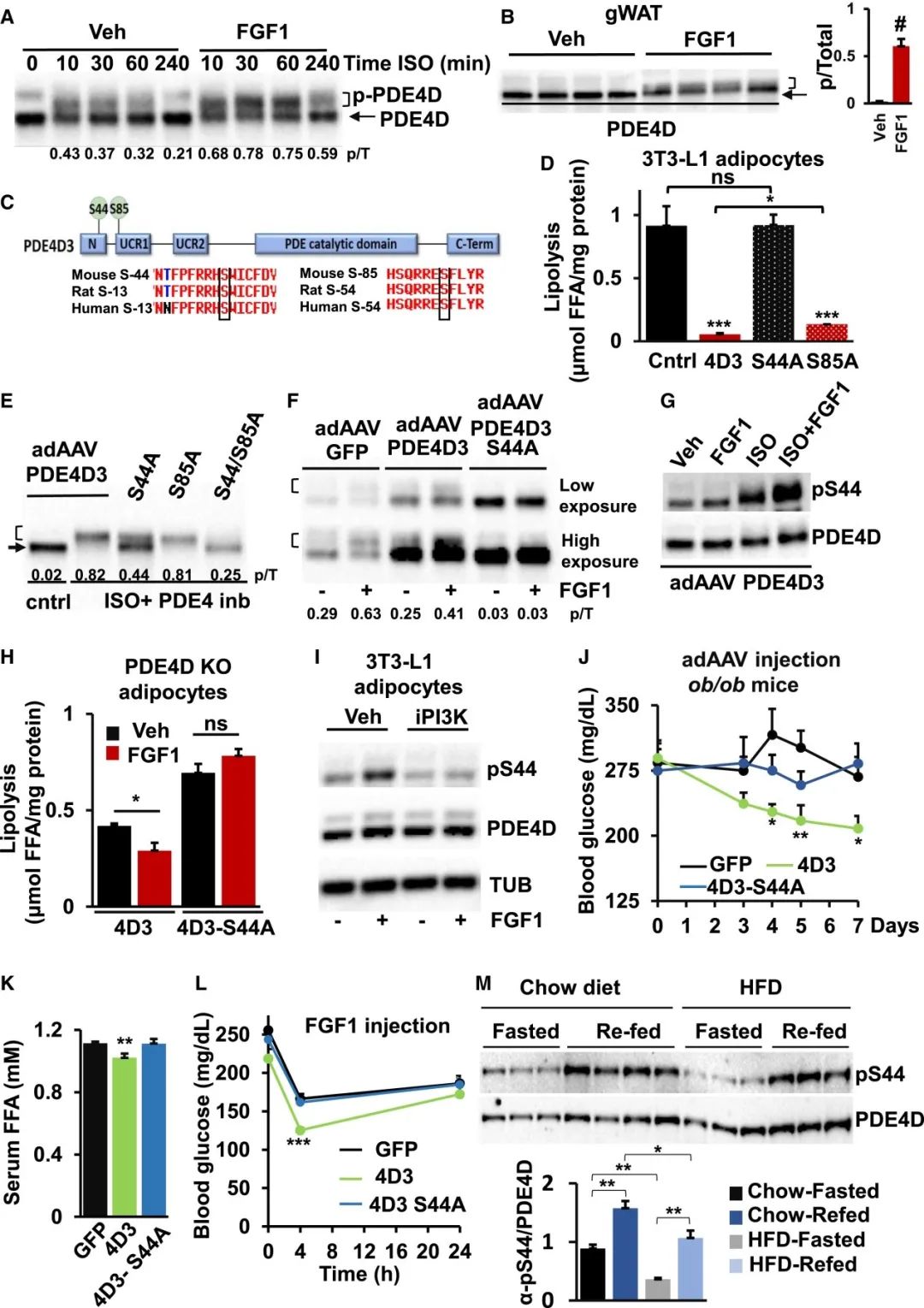
PDE4D3-S44磷酸化是PDE4D3代谢作用所必需的(图源:Cell Metabolism)
总之,研究人员报告了FGF1通过抑制脂肪脂解而显著降低HGP。胰岛素通过脂肪PDE3B抑制脂解,而FGF1依赖PDE4D,使FGF1/PDE4D通路在胰岛素抵抗下保持功能。最后他们进一步确定了Ser44是FGF1诱导PDE4D磷酸化的调节位点,受饱食-空腹循环的调节。
文章作者Gencer Sancar表示,这种机制基本上是第二个循环,具有平行胰岛素途径的所有优点。在胰岛素抵抗中胰岛素信号会受损,然而如果有不同的信号级联,假如其中一个不起作用,则另一个还可以。这样你仍然可以控制脂肪分解和血糖调节。
该研究的联合资深作者Michael Downes说:“FGF1在胰岛素抵抗糖尿病小鼠中诱导持续降低血糖的独特能力是糖尿病患者的一种有前途的治疗途径。我们希望了解这一途径将为糖尿病患者带来更好的治疗。”
参考资料:
[1]Sancar G, LiuSH, Gasser E, et al. FGF1 and insulincontrol lipolysis by convergent pathways [J]. Cell Metabolism 2021, 34,171-183.
DOI: 10.1016/j.cmet.2021.12.004

