Cell Res:清华大学贾怡昌等合作发现肌萎缩性侧索硬化症的一个潜在致病新机理
时间:2023-05-06 15:28:01 热度:37.1℃ 作者:网络
虽然阴离子通道在肌浆网/内质网(SR/ER)中具有活性,但其分子特性和功能尚不清楚。
2023年5月4日,清华大学贾怡昌、中国科学院上海药物研究所高召兵及北京大学樊东升共同通讯在Cell Research在线发表了题为“Disruption of ER ion homeostasis maintained by an ER anion channel CLCC1 contributes to ALS-like pathologies”的研究论文,该研究将氯通道CLIC样1 (CLCC1)的罕见变异与肌萎缩侧索硬化症(ALS)样病理联系起来。该研究通过将纯化的CLCC1结合到脂质双分子层中,证明CLCC1是内质网阴离子通道的成孔成分。CLCC1的消耗减少了ER Ca2+ 的释放,可能是通过反离子机制。
该研究在中国肌萎缩性侧索硬化症(ALS)队列中发现了CLCC1罕见变异。疾病相关的非同义突变损害CLCC1通道传导,并促进突变敲入小鼠脑和脊髓中的错误折叠蛋白积累。在CHaT阳性的运动神经元细胞中,有条件地去除CLCC1会自主地导致泛素阳性包涵体和TDP-43的错位,这是ALS和运动神经元的损失的病理标志。因此,该研究揭示内质网阴离子通道维持内质网离子稳态的失调是内质网未折叠蛋白反应(UPR)和神经退行性疾病病因的基础。

尽管Cl– 是活细胞中最丰富的阴离子,但直到CLC的氯离子通道家族和囊性纤维化跨膜电导调节剂(CFTR)被克隆并将其功能障碍与人类疾病联系起来,人们才对氯离子电流及其功能意义进行了充分的研究。除了细胞表面的通道外,长期以来人们一直认为在胞内膜结合细胞器中也存在Cl– 通道。然而,先前假设的细胞内Cl– 通道,如氯离子通道Ca2+ 激活(CLCAs)和氯离子细胞内通道(CLICs),现在被认为不太可能作为阴离子通道发挥作用。因此,细胞器阴离子通道的分子特性和功能,包括在肌浆网/内质网(SR/ER)中的阴离子通道,在很大程度上仍然是未知的。
作为主要的内部Ca2+ 储存,SR/ER中的Ca2+ 释放由两个阳离子通道介导,即Ryanodine受体(RyRs)和肌醇1,4,5-三磷酸受体(IP3Rs)。在释放过程中,SR/ER膜在Ca2+ 外排时带电,这阻碍了Ca2+ 的持续释放。先前报道的细胞内三聚体阳离子通道(TRICs)平衡SR/ER因释放而损失的正电荷。除阳离子外,阴离子也被提出作为释放的反离子,并且在微粒体制备中已经证明了各种Cl通道的活性。
先前一项使用小鼠正向遗传学的研究表明,氯通道CLIC样1 (CLCC1),氯通道CLIC样1 (CLCC1),一种内质网居住蛋白的缺失,导致内质网应激和神经变性。然而,尽管名字如此,CLCC1与CLIC家族成员或任何已知离子通道的序列相似性很小。此外,CLCC1过表达细胞制备的微粒体中记录的氯离子电流是否真的是由CLCC1介导的问题仍然存在。因此,需要进一步的证据来了解CLCC1是否作为阴离子通道发挥作用。
CLCC1功能丧失介导的ALS和运动神经元丧失(图源自Cell Research )
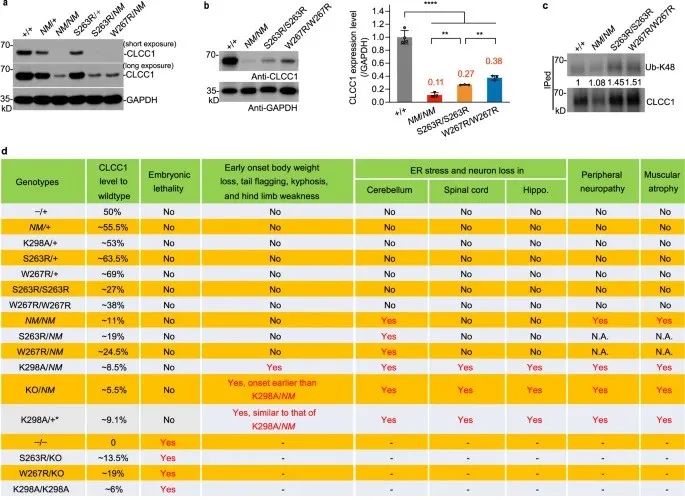
该研究将氯通道CLIC样1 (CLCC1)的罕见变异与肌萎缩侧索硬化症(ALS)样病理联系起来。该研究证明CLCC1是ER阴离子通道的成孔组分,ALS相关突变损害通道电导率。CLCC1形成同聚体,其通道活性被腔内Ca2+ 抑制,但被磷脂酰肌醇4,5-二磷酸(PIP2)促进。该研究发现CLCC1 N端的保守残基D25和D181负责Ca2+ 结合和腔内Ca2+ 介导的通道打开概率抑制, CLCC1腔内环中的K298是关键的PIP2感应残基。研究结果表明CLCC1维持[Cl–]ER和[K+]ER稳态,ER形态及调节ER Ca2+ 稳态。
ALS相关的CLCC1突变形式增加[Cl–]ER稳态并损害ER Ca2+ 稳态。通过多种CLCC1功能缺失等位基因的表型比较,包括ALS相关突变,揭示了体内疾病表型严重程度的CLCC1剂量依赖性。与CLCC1罕见变异在ALS中占主导地位类似,10%的K298A杂合小鼠出现了ALS样症状,这表明由功能丧失突变显性负性诱导的通道病变机制。CLCC1细胞的条件性敲除自主导致运动神经元丢失和内质网应激,错误折叠蛋白积累,以及脊髓的特征性ALS病理。因此,该研究结果支持CLCC1维持的ER离子稳态,该稳态的破坏有助于ALS样病理。
原文链接:
https://www.nature.com/articles/s41422-023-00798-z

