晶泰科技方磊团队JCIM论文:结合人工智能和计算化学方法发现靶向PI5P4K-β的新型活性苗头化合物
时间:2023-09-25 14:12:37 热度:37.1℃ 作者:网络
近年来,计算机辅助药物设计(CADD)和人工智能驱动的药物设计(AIDD),在药物发现和设计等方面中的应用越来越广泛。在此背景下,晶泰科技开发了智能化自动化创新药物发现平台ID4Inno,包含两大计算系统:高精度计算化学平台 ID4Gibbs 和人工智能药物发现平台 ID4Idea,涵盖了智能计算、自动化实验以及专家经验三大模块,该平台以更高的效率、更低的成本探索更广阔的化学空间,大大减少了湿实验的数量,加速了药物发现Design−Make−Test−Analyze(DMTA)循环。
近期,晶泰科技方磊团队在美国化学会出版的计算化学核心期刊Journal of Chemical Information and Modeling上发表了题为 “Hit Identification Driven by Combining Artificial Intelligence and Computational Chemistry Methods: A PI5P4K-β Case Study”的研究论文【1】。该团队在ID4Inno药物发现平台上开发了一种结合人工智能和计算化学来识别苗头化合物的工作流程,该方法具有人工智能的创新性、计算化学的高效性和云计算的高性能等功能特征,并在寻找靶向PI5P4K-β的抗癌药物方面得到了很好的验证。
基于ID4Inno平台的首创苗头化合物发现流程如图-1所示。首先是虚拟筛选(VS):先利用分子对接快速预测靶标与小分子的结合模式并用打分函数评估其亲合性;然后基于半经验量化方法PM7计算的结合能进行筛选,以提高VS阳性率;接着采用两轮MM/GBSA计算筛选出结合能较低的化合物;最后通过人工检查,选择具有合理构象和相互作用模式的化合物进行绝对结合自由能计算,并根据计算结果确定116个候选化合物。考虑到后续结构修饰的合成难度,作者购买了17个化合物并进行生物实验验证。测试结果显示,9个化合物在20 μM时表现出高于50%的抑制率,筛选成功率达53%。其中活性最高的化合物vs1,半抑制浓度为0.80 μM(图-2)。
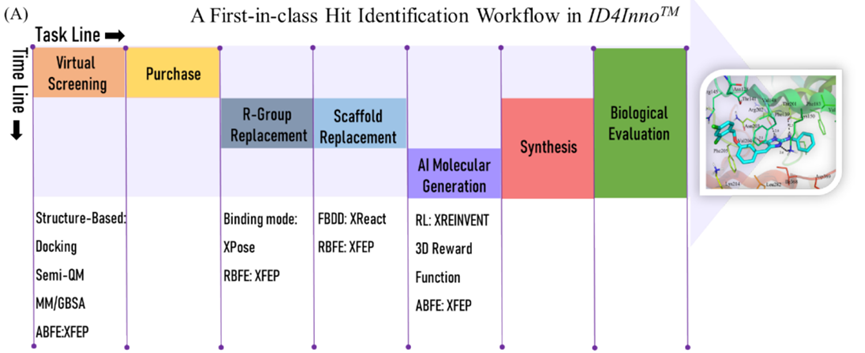
图-1: 基于ID4Inno平台的首创苗头化合物发现流程。
基于XPose预测的vs1与PI5P4K-β的结合模式,作者对vs1应用R基团替换策略设计了一系列小分子,并用XFEP方法预测它们与靶蛋白的结合能力。随后合成了部分化合物并测试它们对PI5P4K-β的酶抑制活性。经构效关系分析,发现在环区进行结构优化后得到的化合物在活性上与vs1相似,XFEP预测的相对结合自由能可以有效指导在远离溶剂区口袋内部的子结构优化(图-3)。
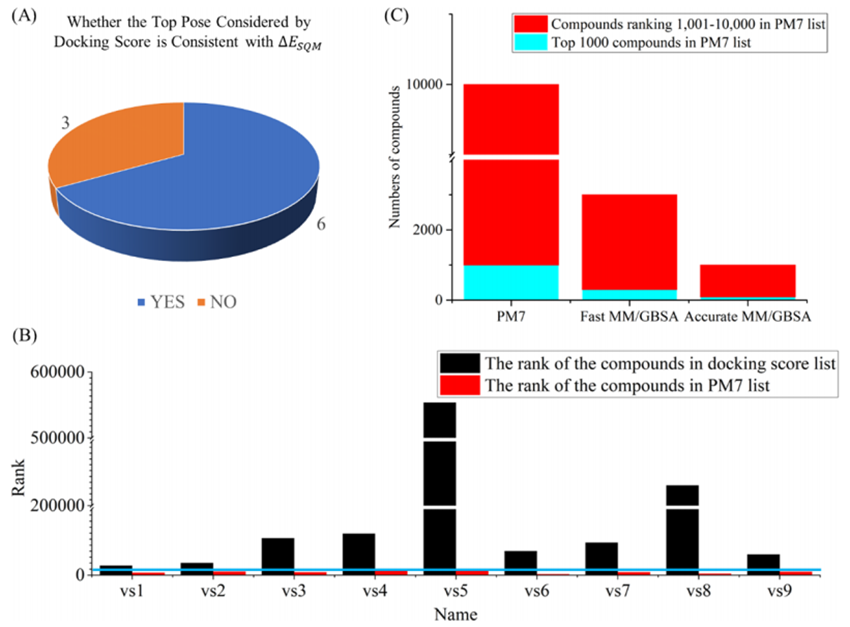
图-2:(A)ΔESQM和对接打分最高结合模式的相关性比较。(B)通过对接打分和ΔESQM对阳性化合物最高结合模式进行排名。(C) PM7前10000个化合物在MMGBSA步骤和PM7步骤的分布比较。
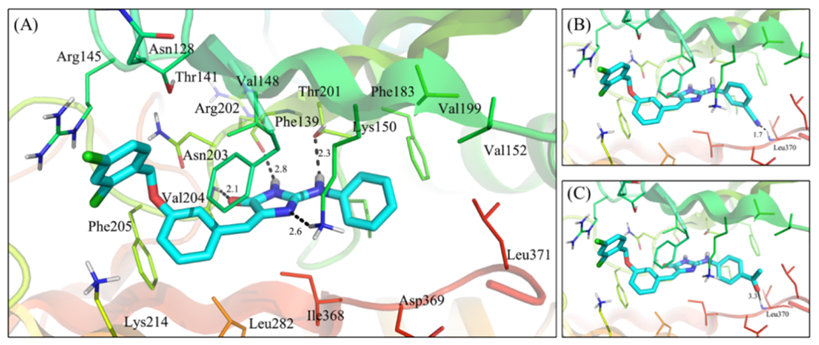
图-3: (A)由XPose预测的vs1的结合模式。(B)d5的结合模式。(C)d8的结合模式。
考虑到XPose结合模式预测和XFEP亲合性预测在环状区域的指导意义较弱,作者采用ID4Idea模块的内部方法XReact对该区域进行结构生成。选择苯胺咪唑酮作为核心骨架(图-4),取代基团仅限于25个重原子并包含1-3个环。在所生成的分子中,对配体效率小于-0.3的分子进行分类,所有类别中每对分子之间的Rogers- Tanimoto相异距离均不大于0.15。从每一类化合物中选择一个化合物进行基于分子动力学模拟的稳定性验证。然后经人工检查、绝对结合自由能过滤等步骤,最终挑选并合成了4个化合物,其中3个化合物的IC50值在可接受范围内。该方法成功地将新型骨架结构空间扩大到了12个化合物。
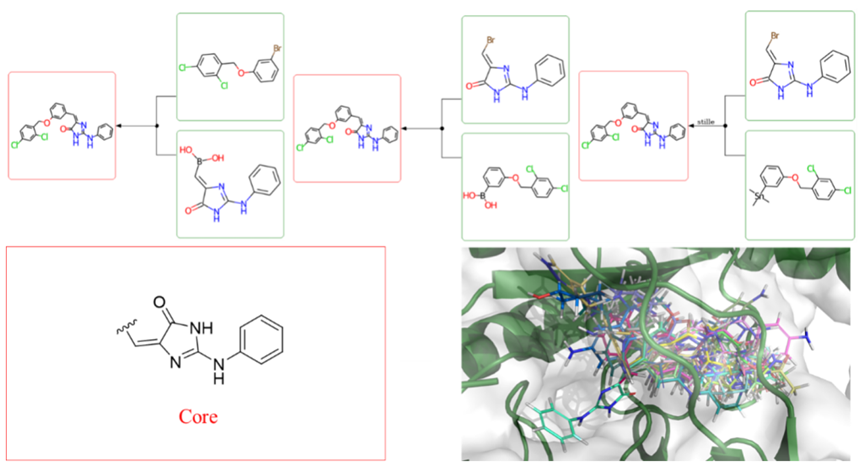
图-4: XReact流程示例。
为了探索更多的活性结构空间,作者应用ID4Idea模块的XREINVENT进行强化学习以生成新分子。该方法所需的预训练生成模型(称为先验模型)是在ChEMBL数据集上训练所得,它具有类似于REINVENT的生成能力,并有可能从相当大的化学空间中采样化合物。在这个先验模型上使用更小的化合物集进行300步迁移学习,然后基于多参数目标进行500步强化学习。强化学习过程选择三维药效团约束评分函数作为奖惩指标。作者使用XREINVENT设计了两轮分子生成,第一轮强化训练中仅考虑药效团模型和分子量,第二轮则增加化合物vs1的子结构作为限制以产生更多类似vs1的分子结构。最终生成的5594个分子结构中有效分子达99.41%,新颖结构占86.5%。使用上述虚拟筛选流程对所有生成分子进行过滤,最终合成并获得两个全新骨架结构,它们对PI5P4K-β的抑制活性小于20 μM。
综合上述虚拟筛选、R基团取代和分子生成等策略,经过生物实验验证最终获得涵盖14个新骨架的26个靶向PI5P4K-β的苗头化合物(IC50<20 μM),其中化合物d5的活性最高,IC50值达到350 nM。

图-5:14个靶向PI5P4K-β活性化合物的化学结构。
小结:
晶泰公司开发的ID4Inno作为一个智能算法、机器人实验和专家经验三位一体的智能化药物发现平台,为新药研发提供个性化的一站式计算服务。(https://www.xtalpi.com/solution)。该论文作者基于ID4Inno平台形成了结合人工智能和计算化学的工作流程,节省了发现活性苗头化合物的时间和成本,彰显了人工智能方法在新药研发中潜在的应用价值。
参考文献:
【1】Wei L, Xu M, Liu Z, et al. Hit Identification Driven by Combining Artificial Intelligence and Computational Chemistry Methods: A PI5P4K-β Case Study. J Chem Inf Model. 2023, 63(16), 5341-5355.

