Nat Genet:单细胞多组学分析揭示慢性炎症是TP53突变白血病进化的驱动因素
时间:2023-10-10 11:24:15 热度:37.1℃ 作者:网络
肿瘤蛋白53(TP53)是人类癌症中最常见的突变基因。TP53突变还与拷贝数改变(CNA)和结构变异密切相关,反映了p53在维持基因组完整性中的作用。骨髓增生性肿瘤(MPN)通过JAK/STAT信号通路基因(JAK2、CALR或MPL)突变在造血干细胞(HSC)中产生,导致骨髓谱系的异常增殖。在约20%-35%MPN进展为继发性急性髓系白血病(sAML)的患者中检测到的TP53突变(统称为TP53-sAML),通常与剩余的p53等位基因的丢失和多个CNA有关。此外, Trp53缺失与JAK2 V617F突变相结合会导致小鼠出现高渗透性髓系白血病。
虽然TP53突变在MPN转化中的作用已确定,但16%的慢性MPN(CP-MPN)中也存在TP53突变亚克隆,并且在大多数情况下,这并不预示着TP53-sAML的发生。此外,人们对获得TP53突变后克隆进化的其他遗传和非遗传决定因素知之甚少。要解决这个问题,需要揭示肿瘤内的多层异质性,包括可靠地鉴定TP53突变、WT等位基因的缺失和CNA的存在。将这种突变图谱与细胞表型和转录特征结合起来,将解决TP53-sAML中白血病干细胞的异常造血分化和分子特性。
近期,来自英国牛津大学联合法国国家卫生及医学研究所的研究人员,通过TARGET-seq技术,将造血干细胞/祖细胞(HSPC)的分子和表型分析与等位基因分辨率突变检测相结合,分析了多层肿瘤内异质性,证明了迄今未被了解的慢性炎症效应,即抑制了TP53 WT HSPC,同时增强TP53突变细胞的适应性优势并促进了遗传进化。相关研究结果发表在Nature Genetics上,文章题为“Single-cell multi-omics identifies chronic inflammation as a driver of TP53-mutant leukemic evolution”。

TP53白血病转化中的趋同克隆进化
为了描述TP53-sAML的遗传图谱,研究人员通过批量靶向NGS和单核苷酸多态性(SNP)阵列分析了33名TP53-sAML患者。在28名患者中检测到了MPN驱动突变(JAK2和CALR),在24名患者中检测到了共同发生的髓系驱动突变。三分之一的患者存在多重TP53突变,其中2例患者存在3个TP53突变;82% TP53单一突变的患者表现出>50%的高变异等位基因频率(VAF)。所有分析的患者均存在CNAs,87%具有复杂核型(≥3个CNAs)。影响TP53位点的缺失或杂合性缺失在43%的患者中可检测到。综上所述,以上发现支持TP53-sAML与复杂的肿瘤内遗传异质性相关。
为了表征肿瘤系统发育和亚克隆结构,研究人员对来自14名TP53-sAML患者的17517个Lin-CD34+造血干细胞/祖细胞进行了TARGET-seq分析,该技术可以对同一单细胞进行等位基因的基因分型、全转录组和免疫表型分析。另外还有9名年龄匹配的健康供体(HD)和8名先骨髓纤维化(MF)患者(图1a)。通常,显性基因通过四种克隆进化模式形成TP53的丢失(图1b-e)。
使用单细胞转录组的CNA分析表明,所有TP53 multihit克隆都至少含有一种高度克隆显性的CNA,其中极少数TP53突变细胞没有CNA证据,另外14名患者中的5名也显示出细胞遗传学上不同的亚克隆(图1f和1g)。为了确认优势HSPC克隆是功能性LSC,研究人员为两名TP53-sAML患者建立了患者来源的异种移植物(PDX)(图1h)。小鼠在27-31周内患上白血病,骨髓中存在大量人CD34+髓样母细胞,其祖细胞表型、TP53突变和CNA与患者的显性克隆相似(图1i)。总之,TP53-sAML中的显性白血病克隆总是以影响TP53的multiple hit为特征,表明WT TP53完全缺失的强大选择压力,加上CNA的获得和复杂的细胞遗传学进化,具有正常核型的TP53 multihit细胞非常少(图1j)。
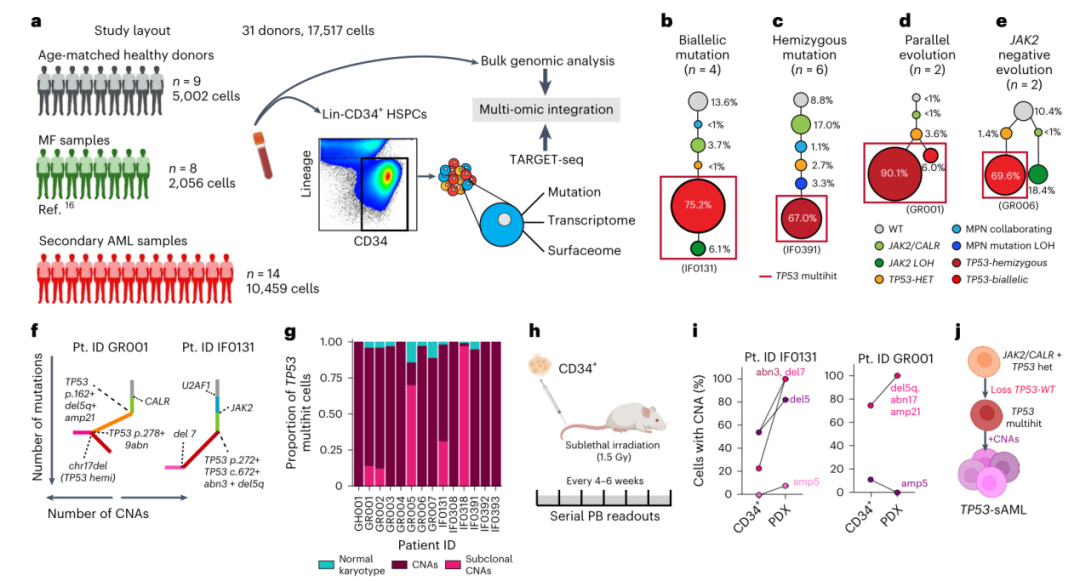
图1. TP53-sAML的克隆进化。
炎症促进TP53相关克隆优势
为了解与白血病进展相关的转录特征,研究人员分析了5名随后发展为TP53-sAML(pre-TP53-sAML)的CP-MPN患者,以及6名携带TP53突变克隆但仍处于CP状态的CP-MPN患者(CP-TP53-MPN)的样本(图2a)。在3名pre-TP53-sAML患者和所有4名CP-TP53-MPN患者中鉴定出杂合TP53克隆(图2b)。来自pre-TP53-sAML患者的TP53杂合HSPC显示TNFα和TGFβ相关基因特征下调,已知这两种基因都与HSC损耗相关,同时氧化磷酸化、DNA修复和干扰素(IFN)响应基因的表达上调(图2c-e)。
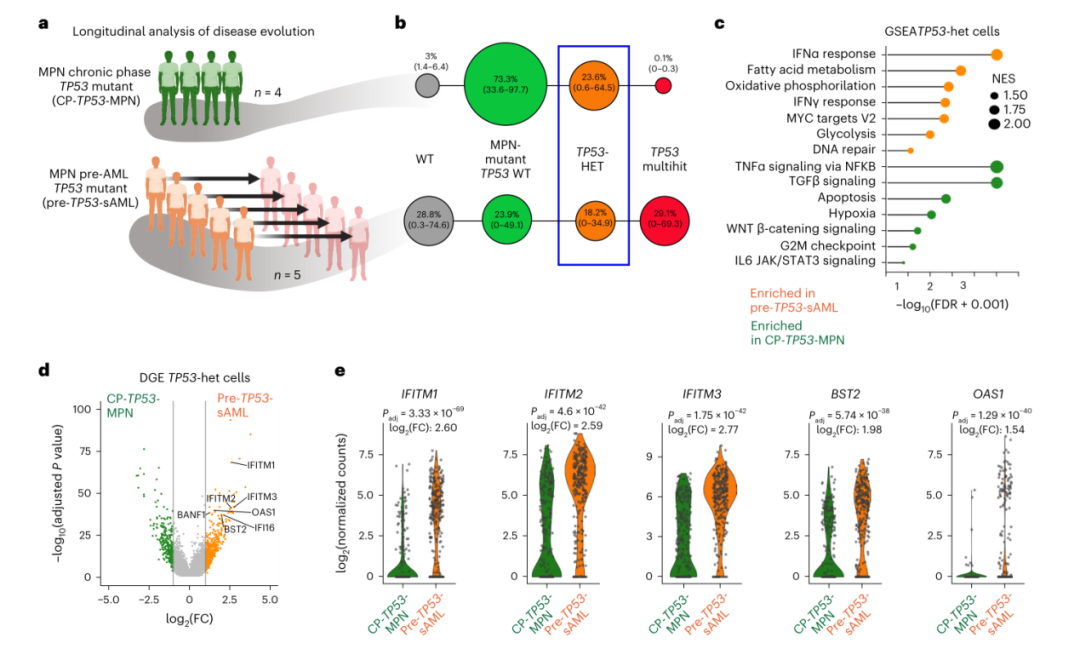
图2. TP53突变型HSPC在转化前炎症通路上调。
为了评估炎症在TP53驱动的白血病进展中的作用,研究人员在CD45.1+Vav-iCre Trp53R172H/+和CD45.2+ Trp53+/+ BM细胞之间进行了竞争性小鼠移植实验,然后重复进行聚肌胞苷酸或脂多糖(LPS)腹腔注射,通过诱导多种促炎细胞因子来概括慢性炎症,已知这些细胞因子在MPN患者的血清中增加,包括IFNγ(图3a)。Trp53突变外周血(PB)骨髓细胞、BM HSC(Lin-Sca1+c-Kit+CD150+CD48-)和LSK(Lin-Sca1+c-Kit+)在聚肌胞苷酸处理后选择性富集(图3b、3c)。至关重要的是,Trp53突变体HSC和LSK的适应性优势是通过Trp53R172H/+ HSPC数量的增加和WT竞争细胞数量的减少来发挥的(图3d、3e)。用LPS治疗嵌合小鼠(图3a),通过释放IL1β和IL6等诱导炎症反应,导致Trp53突变的PB骨髓细胞和LSK数量类似增加(图3f、3g),这些结果表明多种炎症刺激可以促进Trp53突变克隆的扩增。
为了确定炎症如何改变造血分化并施加选择压力来驱动Trp53突变克隆的扩增,研究人员建立了诱导型SCL-CreERT Trp53R172H/+小鼠模型(图3h)。与Vav-iCre模型类似,聚肌胞苷酸治疗促进了PB中髓系Trp53突变细胞的选择,其中由PB白细胞中的炎症刺激诱导的髓系偏向与Trp53突变(图3i、3j)。与预期一致,在聚肌胞苷酸治疗后,WT竞争红系祖细胞的数量减少了,而Trp53突变与不受炎症影响的红系祖细胞的增加相关(图3k)。在聚肌胞苷酸处理的WT和Trp53突变的LSK中,细胞周期也同样增加;与WT对应物相比,Trp53突变的LSK对炎症诱导的细胞凋亡具有抵抗力(图3l、3m)。
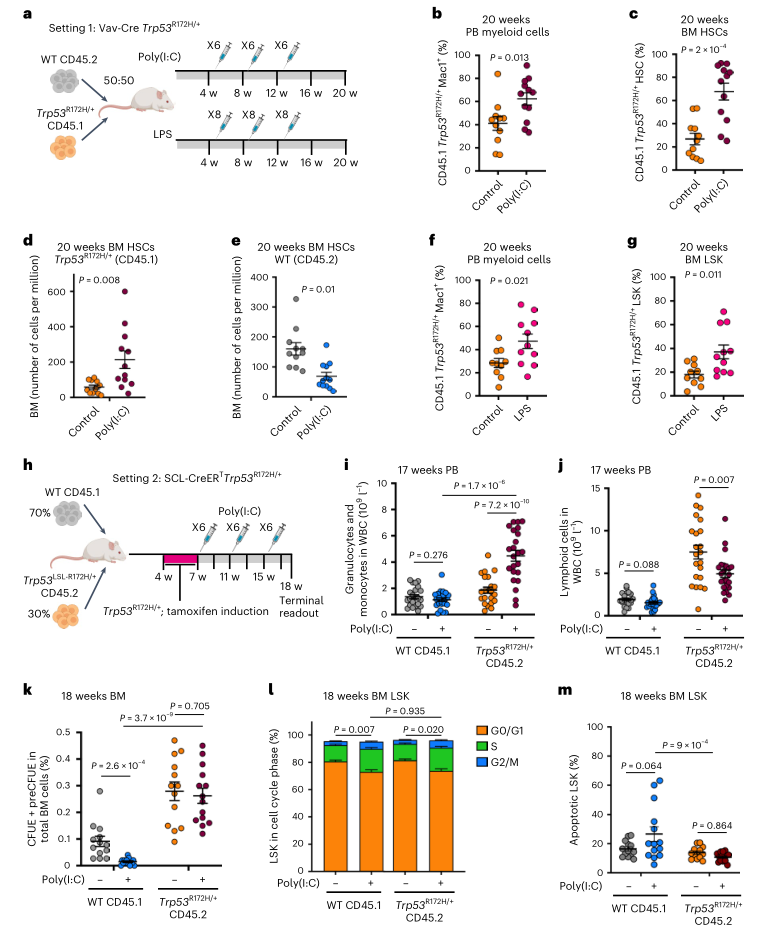
图3. 炎症促进TP53相关克隆优势。
炎症促进Trp53突变HSPC的遗传进化
由于退出休眠会促进DNA损伤诱导的HSPC损耗,研究人员推断Trp53突变可能会拯救由慢性炎症相关增殖应激驱动的DNA损伤(否则会发生细胞凋亡)的HSPC。研究人员对前述的实验样本进行了M-FISH核型分析,发现经过聚肌胞苷酸处理后,Trp53突变的LSK衍生细胞中核型异常的频率和数量显著增加(图4a-d) 。总的来说,以上结果表明慢性炎症促进TP53突变细胞的存活和遗传进化,同时抑制WT造血,最终促进TP53突变HSPC的克隆扩增(图4e)。
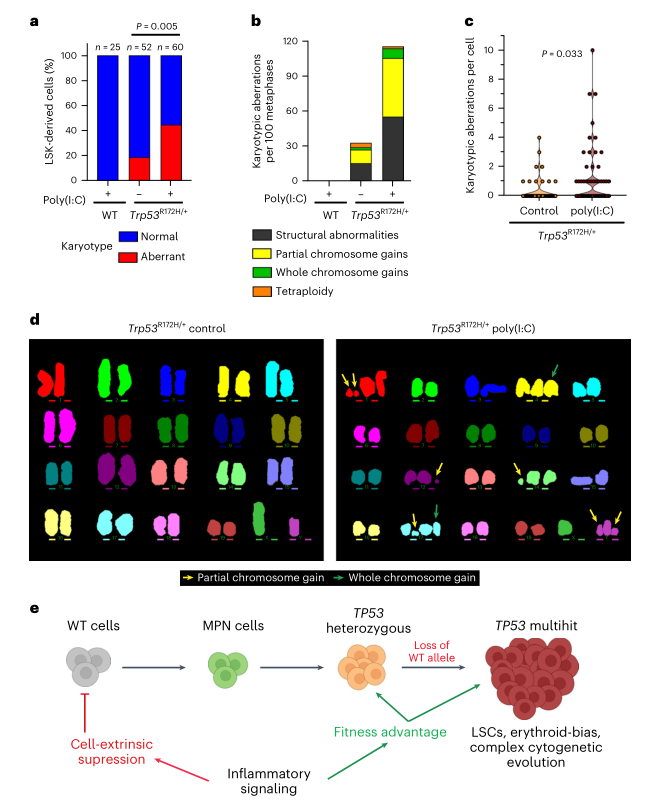
图4. 炎症导致Trp53突变细胞的遗传不稳定。
该研究通过单细胞多组学分析揭示了TP53突变驱动的疾病转化中的多层遗传、细胞和分子瘤内异质性。白血病HSPC的等位基因分辨率基因分型揭示了使TP53产生错义突变、丢失TP53剩余的等位基因和发生复杂CNA的强大选择压力,包括TP53-sAML LSC扩增期间具有平行遗传进化的情况。
该研究结果提供了有关TP53突变相关疾病转化的遗传和非遗传决定因素之间相互作用的重要概念进步。这将有助于制定TP53突变型白血病的早期检测和治疗策略。因为TP53是人类癌症中最常见的突变基因,研究人员预计这些发现将与其他癌症类型具有更广泛的相关性。
参考资料:
Rodriguez-Meira, A., Norfo, R., Wen, S. et al. Single-cell multi-omics identifies chronic inflammation as a driver of TP53-mutant leukemic evolution. Nat Genet 55, 1531–1541 (2023). https://doi.org/10.1038/s41588-023-01480-1

