视神经脊髓炎谱系疾病(NMOSD)
时间:2024-08-14 16:00:54 热度:37.1℃ 作者:网络
论坛导读:视神经脊髓炎谱系障碍(NMOSD)是一个统称术语,指AQP4-IgG阳性的视神经脊髓炎(NMO)及其表现形式,以及一些不含AQP4-IgG的密切相关的临床综合征。NMOSD最初被认为是多发性硬化(MS)的亚变异体,但现在被广泛认为是在免疫发病机制、临床表现、最佳治疗和预后方面与MS不同的疾病。
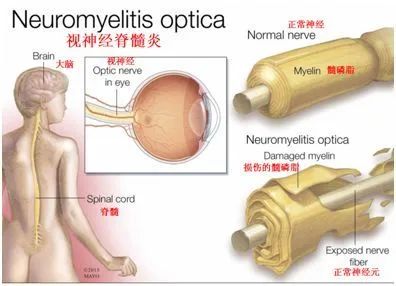
视神经脊髓炎谱系障碍(NMOSD)用于指中枢神经系统(CNS)的炎性疾病,主要影响视神经、脊髓,偶尔影响脑干,引起视神经炎、脊髓炎和脑干脑炎的急性发作。在大多数情况下,NMOSD与针对水通道蛋白-4(中枢神经系统中最常见的水通道)的致病性免疫球蛋白G (IgG)抗体有关。2004/2005年发现的AQP4-IgG对NMOSD的疾病分类、诊断和治疗具有重要意义。NMOSD必须被认为是所有自身免疫性病因的中枢神经系统炎症患者的潜在鉴别诊断,特别是如果他们患有视神经炎、脊髓炎或脑干脑炎,无论性别、年龄和种族。流行病学研究在纳入标准和方法方面存在显著的异质性;特别是,并非所有研究都区分了AQP4-IgG阳性和AQP4-IgG阴性患者。然而,已经发现了一些风险因素。NMOSD已被证明主要影响女性,在一些非高加索人群中更常见,并且在大多数情况下在成年期开始。
NMO既往被认作是多发性硬化(multiple sclerosis,MS)的一个亚型,直到2004 年Lennon 等在NMO 患者血清中发现水通道蛋白4 抗体(aquaporin4 immunoglobulin G,AQP4-IgG),使NMO 与MS 从发病机制上区分开来。2015 年国际NMO 诊断小组(international panel for NMO diagnosis,IPND)首次提出视神经脊髓炎谱系疾病(neuromyelitis optic spectrum disorder,NMOSD)的概念,并且分别制定了AQP4-IgG 阳性或阴性时NMOSD 的诊断标准。
在大多数研究中,中年人的发病率最高(例如,在一项大型欧洲队列研究中,AQP4-IgG阳性患者的年龄约为40岁,AQP4-IgG阴性患者的年龄约为38.5岁)。然而,老年人和儿童也必须认真考虑NMOSD的诊断。在一些(混合AQP4-IgG阳性和AQP4-IgG阴性)队列中,迟发性NMOSD (> 60岁)占所有NMOSD事件病例的20–28 %,该疾病甚至可能仅在高龄(> 75岁)时开始。LO-NMOSD与预后不佳有关。相比之下,多发性硬化(MS)和成人MOG-EM/MOGAD的临床疾病发作的中位年龄约为30岁。NMOSD必须被认为是所有表现为急性横贯性脊髓炎发作和/或(单侧或,少数情况下,双侧)急性NMOSD的两个临床特征的患者的鉴别诊断,但也包括表现为疑似脑炎或脑干脑炎的患者。脊髓炎可同时发生,或更常见的是相继发生。脊髓炎和视神经炎的结合给了这种疾病的名字。
NMOSD是一种主要累及青壮年人群,高复发、高致残性中枢神经系统炎性脱髓鞘病,其发病机制主要与水通道蛋白4(aquaporin-4,AQP4)抗体相关,需要早诊断及长期预防复发治疗。随着我国《中国视神经脊髓炎谱系疾病诊断与治疗指南》(2016版)的颁布,NMOSD的正确诊疗率显著提高。2020年发布了基于住院登记信息的流行病学数据;同时国际上完成了多项针对AQP4-IgG阳性NMOSD的RCT研究,均取得了显著疗效;一些新型作用靶点的单克隆抗体药物在中国食品药品监督管理局递交了申请,已经或即将获批上市;NMOSD 治疗迎来了高效、高循证医学证据治疗的时代。基于对流行病学及新药物循证数据的更新以及MOG-IgG相关疾病(MOG-IgGassociateddisorders)的定义分类,完善疾病管理,中国免疫学会神经免疫分会组织国内专家对NMOSD指南进行了改版更新。
必须对疑似脊髓炎患者进行感觉、运动(包括呼吸)、膀胱/肠道和性功能障碍检查。此外,应该评估疼痛,除了临床检查外,最好使用结构化问卷。疼痛作为脊髓炎的直接或长期后遗症,在AQP4-IgG阳性和AQP4-IgG阴性的NMOSD中都非常普遍,包括神经性疼痛、疼痛性强直性痉挛、神经性瘙痒和痉挛相关疼痛;感觉异常(在胸部脊髓炎的情况下通常为带状)和痛觉过敏/异常性疼痛是常见的。
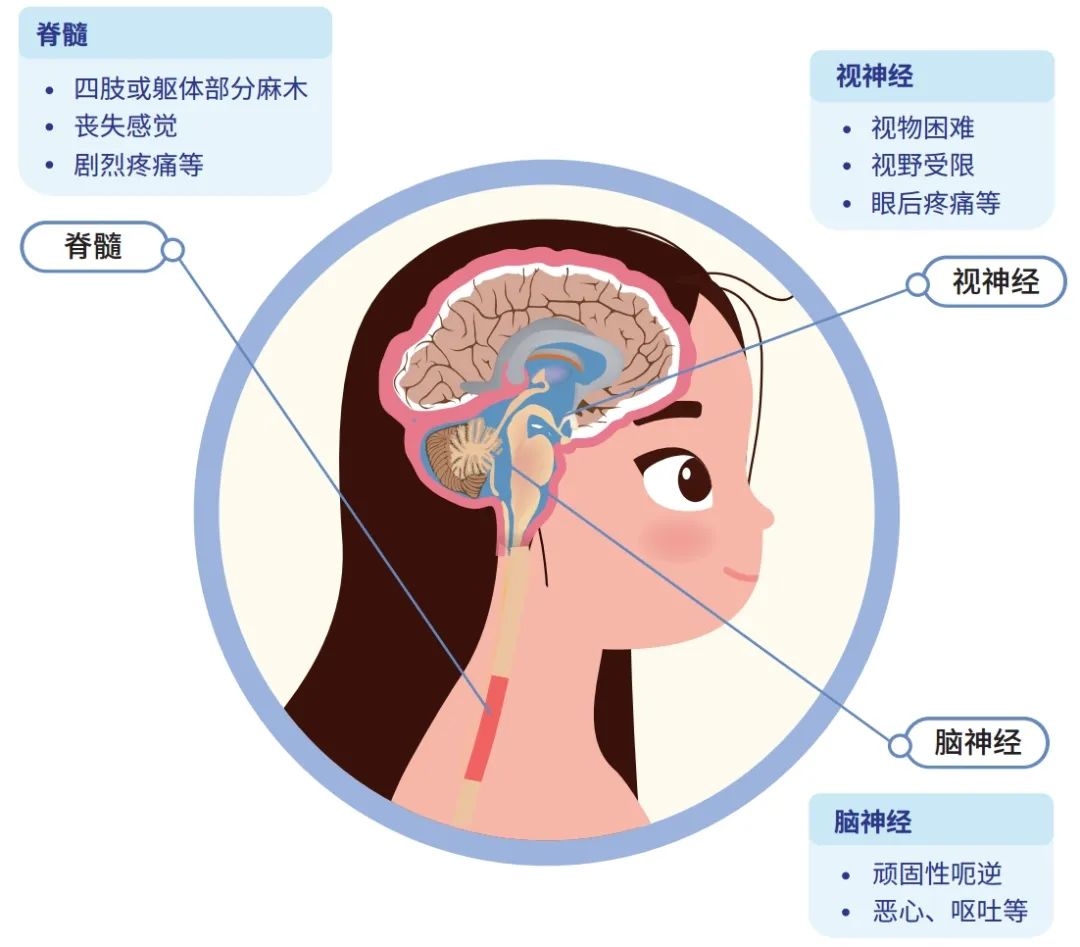
视神经炎是炎症脱髓鞘性病变,是NMOSD 的核心症状之一。通过视神经MRI 可客观观察和评价病变的范围和信号特点。视神经脱髓鞘病灶长度通常大于视神经长度的一半,易常累及视交叉和视神经后段。

急性脊髓炎临床特征为急性或亚急性运动、感觉、自主神经功能障碍,主要表现为截瘫或四肢瘫痪,感觉异常,二便障碍等。长节段横贯性脊髓损害是NMOSD 的特征性表现之一,MRI 矢状位可见脊髓病灶多位于颈胸段,典型LETM 病灶累及上位胸髓,颈髓病灶可向上延伸至延髓,病灶纵向长度往往超过3 个椎体节段,部分病例脊髓病灶可累及脊髓全长,病灶横断面积大于相应节段脊髓截面面积的50%。
脊髓MRI 显示病变主要位于灰质区,部分累及白质,急性期病灶节段脊髓肿胀增粗,T1WI 呈等或低信号,T2WI 高信号,矢状位为斑点状或线样高信号,轴位可见“亮斑征”,T1WI 钆增强扫描病灶强化不规则、均匀或不均匀,呈环形或斑片状;慢性期,病灶T2WI 呈高信号,无明显强化,可出现髓内软化灶,通常伴有边界清晰的局灶性或广泛长节段脊髓萎缩。
极后区(area postrema,AP)位于脑干的延髓背侧面,第四脑室底部,孤束核背侧, 1953 年Borison 等证实了极后区是呕吐反射的化学感受区。极后区综合征(area postrema syndrome,APS)为NMOSD 较常见的核心症状之一,主要的临床表现为不明原因的顽固性呃逆、恶心和呕吐。病灶可由脊髓上段病灶延伸而来,也可仅存在于延髓背侧,MRI 主要表现为延髓背侧斑片样或线样病灶,T1WI 呈等或低信号,T2WI、FLAIR 呈高信号,T1WI 钆增强病灶呈均匀或不均匀斑片状强化,边界模糊,轴位MRI 显示病灶通常以中央管为中心呈对称性分布。APS 作为NMOSD 的首发症状在疾病早期单独存在时,极易被误诊为消化系统疾病,通过影像学检查可早期识别极后区综合征,协助临床医师进行诊断及鉴别诊断。但也有文献报道首发症状为APS 的NMOSD 患者MRI 显示无延髓背侧病灶,推测其可能与患者进行MRI检查的时间有关,提出多次MRI 随访的重要性。
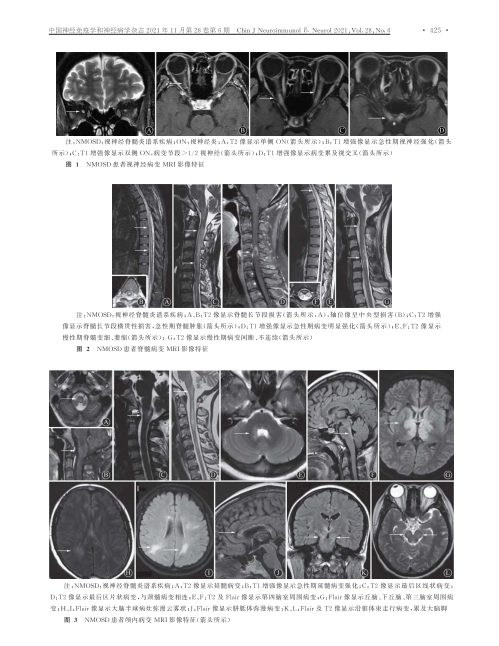
大脑综合征也是NMOSD 的核心症状之一,主要表现为高级皮层功能障碍,具体临床表现与病灶累及部位有关。MRI 表现包括皮质下或深层白质点状病灶或范围较大的融合性片状病灶,病灶呈T1WI 等或低信号,T2WI 和FLAIR 高信号,T1WI 钆增强病灶无强化或部分强化,典型特征为“云雾状”多发斑点样强化,边缘模糊。病变可累及胼胝体(病变范围常大于胼胝体长度的1 /2)、侧脑室周围、基底节,皮质脊髓束病灶为NMOSD 较特异性病灶急性脑干综合征(acute brain stem syndrome,ABS)指NMOSD 患者脑干的损伤,临床表现包括眼球震颤、复视、眩晕、皮肤瘙痒、吞咽困难等。
AQP4-IgG 是具有高度特异性的诊断标志物,特异度高达90%,敏感度约70%。推荐使用基于细胞转染的免疫荧光技术(cell based transfection immunofluorescence assay,CBA)或流式细胞技术进行血清检测。酶联免疫吸附试验(enzyme linked immunosorbent assay, ELISA)较为敏感,但特异度有所降低,不推荐作为确立诊断的检测方法,但纵向监测抗体滴定度对疾病进展和治疗的评估有一定价值。脑脊液( CSF) 压力多数正常;急性期白细 胞多大于 10✖106/L,约1/3患者大于 50✖106/L, 少数病例可达500✖106/L;可 见 中性粒细胞及嗜酸粒细胞增多 。 急性期生化:蛋白多明显增高 ,可大于 1g/L,糖及氯化物多正常;约20%患者CSF 特异性寡克隆区带(OCB)阳性 ,IgG明显增高。
MOG-IgG是MOGAD的生物诊断标志物,几乎不与AQP4-IgG同时阳性,具有重要鉴别诊断价值。推荐采用CBA法对血清及CSF MOG-IgG进行检测。需要注意的是,一些疾病急性期可表现为一过性MOG-IgG阳性,需结合临床进行解读。约近50%AQP4-IgG阳性NMOSD患者合并其他自身免疫抗体阳性,常见有血清抗核抗体(ANAs)抗SSA抗体、抗SSB抗体、甲状腺过氧化酶抗体(TPO)阳性等。神经丝轻链(neurofilament light chain,NfL)作为神经元损伤的生物标记物可在多种疾病中被观察到。尽管其特异度不高,但在动态反映神经元损伤程度上被认为是较好的生物学指标,有利于观察疾病的进展及不可逆性损伤,可以作为NMOSD残障进展和治疗评价的生物学指标,同时需要综合如高血压、糖尿病、脑梗死等合并症因素的共同影响。
NMOSD的诊断及鉴别诊断至关重要,需要注意疾病的复杂性以及检测方法的局限性等因素影响,NMOSD患者首次发作或病程在某一阶段AQP4-IgG检测均可能为阴性。对于早期或临床及影像特征不典型的病例,应该充分完善实验室及其他相关检查,同时与可能疾病相鉴别,并进行动态随访,查找相关支持或排除证据,对合并其他自身抗体阳性患者,如自身免疫性脑炎(autoimmuneencephalitis,AE),需结合临床综合评价哪一个是责任致病抗体,切忌唯抗体阳性诊断。
NMOSD的治疗分为急性期治疗、序贯治疗(预防复发治疗)、对症治疗和康复治疗。NMOSD药物治疗原则:NMOSD任何一次临床发作均有可能带来不可逆性损伤;其残障主要归因于发作后视觉功能缺损的累积。对于AQP4-IgG阳性以及AQP4-IgG阴性复发病程的患者,一经诊断应尽早开始序贯治疗,并坚持长程治疗。NMOSD治疗药物的选择应在遵循循证证据基础上,结合安全性、有效性以及患者意愿进行。长期免疫抑制治疗有增加机会性感染和肿瘤的风险,推荐定期进行安全及有效指标监测,有条件的地区单位可开展免疫抑制剂药物基因筛查及血药浓度监测,做到个体化指导。近年来,一些新兴治疗靶点单克隆抗体药物不断涌现,RCT研究结果显示出显著疗效,为NMOSD治疗领域提供了更高的循证依据。国际上已有 3 种药物被美国FDA或欧盟正式批准用于治疗NMOSD,包括补体抑制剂、IL-6 受体阻断剂以及 B淋巴细胞耗竭剂。2021年4月30日中国国家药品监督管理局正式批准萨特利珠单抗用于治疗 12 周岁以上AQP4-IgG阳性的NMOSD患者,成为中国大陆首个获批 NMOSD 治疗适应证的药物。
急性期治疗目标减轻急性期症状、缩短病程、改善残疾程度和防治并发症。治疗人群:有客观临床及影像发作证据的急性发作期患者。序贯治疗(预防复发治疗)治疗目标预防复发,减少疾病反复发作导致的神经功能障碍累积。治疗人群适用于AQP4-IgG阳性以及 AQP4-IgG未知或阴性、复发病程的NMOSD患者。确诊后尽早启动治疗,并坚持长程治疗。治疗药物分为单克隆抗体药物及免疫抑制剂两大类。应该避免应用的药物: 一些治疗MS的药物,如B干扰素、芬戈莫德、那他珠单抗阿伦单抗可能会导致NMOSD的恶化,不推荐应用。
NMOSD的康复治疗同样重要 。对伴有肢体 、吞咽等功能障碍的患者, 应早期在专业医生的指导下进行相应的功能康复训练,在应用大剂量激素治疗时,避免过度活动,以免加重骨质疏松及股骨头负重 。 当激素减量到小剂量时,可鼓励活动,进行相应的康复训练 。生活中保持心情愉快,戒烟,不饮酒,作息规律,合理饮食,保持适当户外阳光下活动,补充维生素 D等 。
参考文献
黄德晖,吴卫平,胡学强. 中国视神经脊髓炎谱系疾病诊断与治疗指南(2021版).中国神经免疫学和神经病学杂志.2021.28(6):423-136.
Jarius S, et al. Update on the diagnosis and treatment of neuromyelits optica spectrum disorders (NMOSD) - revised recommendations of the Neuromyelitis Optica Study Group (NEMOS). Part I: Diagnosis and differential diagnosis. J Neurol. 2023 Jul;270(7):3341-3368. doi: 10.1007/s00415-023-11634-0.
邹彤,黄靖,卢洁.视神经脊髓炎谱系疾病磁共振研究现状及进展[J].医学影像学杂志,2023,33(07):1244-1247.

