28岁的他突然全身酸痛,竟是基因缺陷引起的罕见病作祟
时间:2024-11-11 06:03:22 热度:37.1℃ 作者:网络
小李是一位28岁的白领,平时身体健康,热爱运动。然而,最近他双手和双脚不明原因地出现剧烈灼痛,尤其在运动或天气炎热时,疼痛尤为强烈。起初,他以为是劳累过度或神经问题所致,但经过检查并未发现异常。随着症状逐渐加重,小李还出现了头晕、乏力、出汗减少等症状。在朋友劝说下终于来到了医院。
接诊医生了解了小李的病情,追问了其家族史发现小李的母亲长期蛋白尿,没有正规就诊;患者舅舅年少时常有发作性四肢肢端烧灼痛,也未正规诊治。医生立马为小李安排了基因筛查,并最终确诊了一种罕见的遗传性疾病——法布雷病。
什么是法布雷病?
法布雷病(FD) 又名“Anderson-Fabry disease“,由英国Anderson和德国Fabry分别于1898年首次报道。其是X染色体连锁的遗传性疾病,为位于X染色体的GLA基因突变,导致其编码的α-GalA活性降低或完全缺乏,其代谢底物三己糖酰基鞘脂醇(GL-3)及其衍生物脱乙酰基GL-3(Lyso-GL-3)不能及时被降解,在肾脏、心脏、神经、皮肤等大量贮积,进而引起一系列的临床症状。我国已于2018年将法布雷病列入首批罕见病目录。
临床表现
FD根据发病时间和临床表现的不同分为经典型和迟发性,迟发型较经典型常见,约为后者的10倍。迟发型FD多见于女性,常于40岁后发病,主要累积心脏和肾脏。经典型FD多见于男性患者,多于儿童期开始发病,主要以周围神经系统及周围组织器官受累起病,如神经性疼痛、眩晕,皮肤出现血管角质瘤等,晚期开始出现肾脏、心脏及脑部的病变,如血尿、蛋白尿、心肌肥厚、心律失常、心脏瓣膜病、脑白质变性、TIA等。
诊断
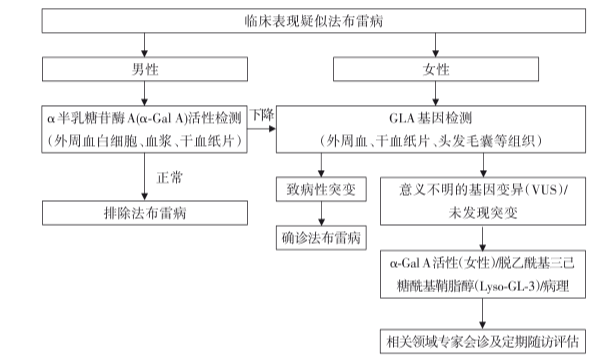
1.α-GalA活性检测:男性患者α-GlA活性严重下降或缺失,可提示患有法布雷病。女性患者受X染色体随机失活的影响,α-GalA活性水平不一,60%以上的女性患者α-GlA活性在参考值范围内。
2.基因检测:经典型常见无义突变、剪切突变和大多数移码突变,迟发型常见错义突变和罕见剪切突变。但仅有80%左右患者可检测到致病基因突变或良性基因突变。
3.生物标志物检测:
(1)血浆GL-3水平:是诊断法布雷病常用的生化指标,男性患者血浆GL-3水平明显高于健康人群,而女性患者普遍较低,且多处于参考值范围。
(2)血浆Lyso-GL-3水平:其敏感度比GL-3更高,且与临床表型有良好的相关性。血浆Lyso-GL-3水平的显著升高有助于区分经典型和迟发型。
4.组织病理学活检:具有辅助诊断意义,可检测心脏、肾脏、皮肤、或神经组织。
治疗
1.酶替代治疗(ERT):目前,ERT药物包括阿加糖酶β和阿加糖酶α。ERT可通过外源性补充基因重组的α-GlA,替代患者体内酶活性降低或完全缺乏的α-GlA,促进GL-3的分解,减少GL-3和Lyso-GL-3在器官组织的贮积,减轻患者疼痛程度,减少蛋白尿,并改善其他相应症状,阻止或延缓多系统病变发生。
2. 对症治疗:法布雷病累及多个组织器官,对症治疗主要是针对各脏器受累情况(具体治疗方案需专科医生评估)。
3.其他治疗:分子伴侣疗法是一种口服小分子药物,可选择性地、可逆性地与结构、功能有缺陷的α-GlA结合,稳定蛋白构象,帮助蛋白正确折叠以发挥正常功能。目前临床使用较多的是米加司他,需注意的是,米加司他仅适用于部分错义突变的法布雷病患者。
此外,底物减少治疗、基因治疗及基于mRNA治疗等一些新的药物或治疗策略正在临床试验或研发中,有望为法布雷病的治疗提供新的方向。
诊断流程图
参考资料:
1. 中国法布雷病专家协作组. 中国法布雷病诊疗专家共识(2021年版). 中华内科杂志,2021,60(04):321-330. DOI:10.3760/cma.j.cn112138-20201218-01028
2. 法布雷病全国专家协作组,中国医药教育协会临床肾脏病学专业委员会. 法布雷病多学科联合全程管理路径. 中华内科杂志,2023,62(08):949-955. DOI:10.3760/cma.j.cn112138-20230218-00095

