脊髓性肌萎缩症:诊疗与预防|指南荟萃
时间:2023-05-19 09:49:11 热度:37.1℃ 作者:网络
脊髓性肌萎缩症(spinal muscular atrophy,SMA)是最常见的婴幼儿常染色体隐性遗传病之一,主要是运动神经元存活基因1突变导致的脊髓前角运动神经元退行病变,以肌无力和肌萎缩为主要临床表现。由于它会造成运动神经元退化、肌肉萎缩,肌肉无力,最终造成死亡。我国SMA致病变异的总体人群携带率为1.2%~2.2%,发生出生缺陷的风险大。近年来,得益于多学科管理、疾病修正治疗药物和医保政策等方面的提升,我国SMA患者的生存期及生活质量在整体上得到大幅改善。然而,年龄较大的青少年及成人患者因其病程长,患者状况错综复杂,因运动功能逐渐下降造成身体残疾后,会相应出现骨骼、呼吸、吞咽及营养、心理等多系统的损害,针对这部分患者的精准诊疗和多学科管理仍存在未尽之需。

SMA中最常见的类型是运动神经元存活基因1 ( SMNI)突变所导致的常染色体隐性遗传病,即5号染色体长臂突变导致的5qMA,发病率约为1/10 000,人群携带率约为1 /50。SMA表型复杂,根据发病年龄及所获得的最大运动里程碑不同,分为5个亚型(0~4型)。大约20%~30%的SMA为II型。SMA-II型的患者通常在7~18个月内出现全身无力,在无帮助的情况下无法坐或站立。手指姿势震颤、肌肉骨骼畸形、腓肠肌假性肥大或呼吸衰竭也可能出现。尽管SMA类型的数量因分类而异,但最常的类型是I~IV,其中I~III型(少年型)是最常见的形式。症状发作年龄最早的为SMA-I型,也是最常见和最严重的类型,且死亡率最高。SMA-III型是病情最轻的少年型,但 SMA-IV型是所有类型中最轻、最良性的类型。
人类与SMA这一种严重致死致残疾病的斗争已经有百余年的历史。1891年,奥地利神经病学家第一次描述记录了1型SMA的症状。随着进入20世纪,人类对于SMA疾病发病机制的不断研究和破解,SMA不再成为无药可治的绝症。迄今为比,共有3种SMA的疾病修饰治疗药物(DMTs)上市,包括反义寡核苷酸药物Nusinersen、小分子药物Risdiplam 及基因替代治疗药物Zolgensma ,但目前缺乏在3种药物中进行疗效及安全性的平行对照的临床研究。

三级预防的策略可有效降低SMA的发生率。为了进一步规范我国的SMA三级预防,由中华医学会神经病学分会神经遗传学组牵头国内多学科专家,遵循国内外指南的撰写规范,根据我国的研究现状和患者的特点,参考国际上最新的研究证据,并借鉴相关指南,最终总结出适合我国国情的SMA三级预防指南。该指南的颁布将为我国SMA的预防发挥积极作用,具有重要的现实意义,并将会产生深远的影响。
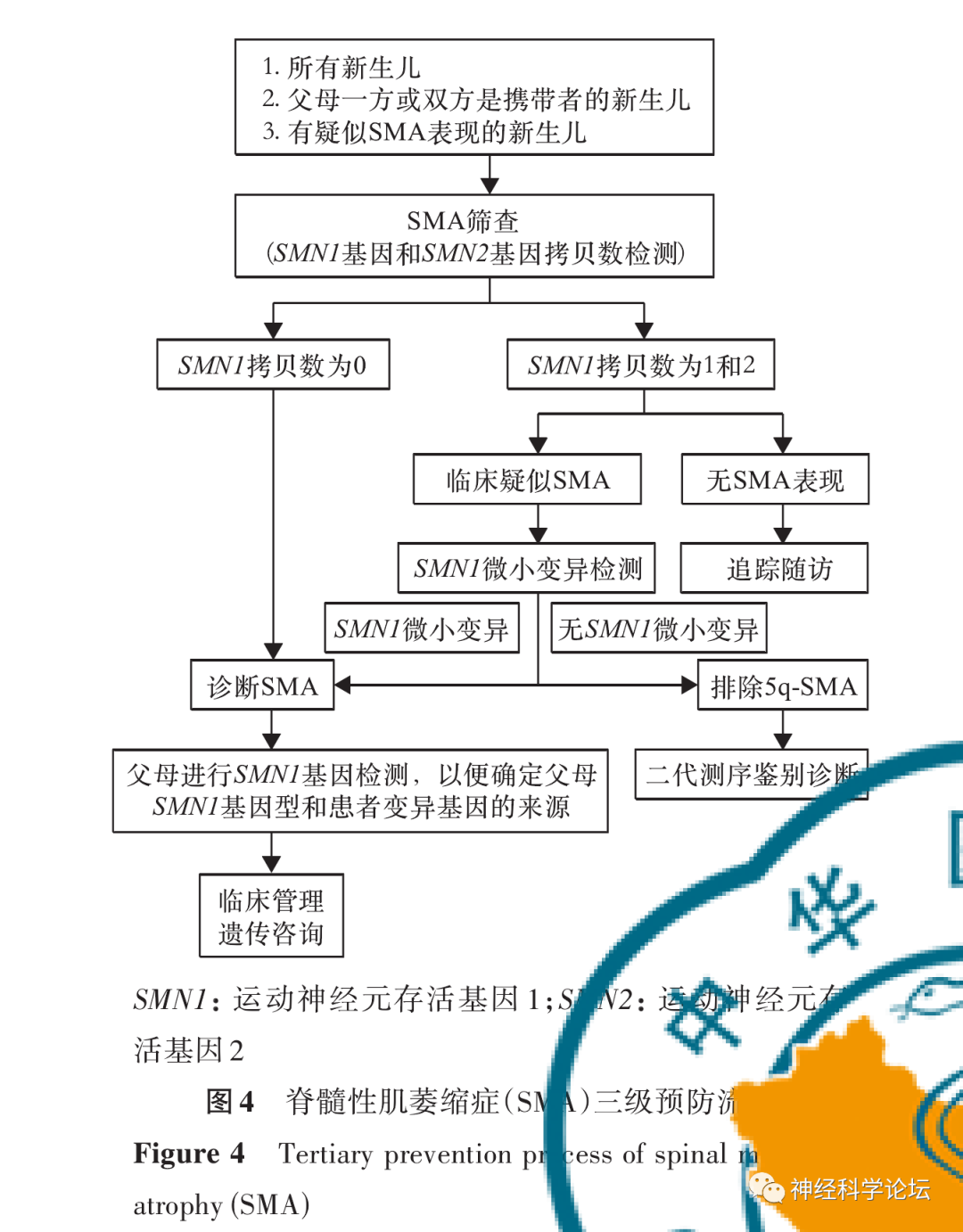
三级预防 (Tertiary Prevention)亦称临床预防。三级预防可以防止伤残和促进功能恢复,提高生存质量,延长寿命,降低病死率。主要是对症治疗和康复治疗措施。由于三种预防措施是连续的梯次性预防措施,因而称之为三级预防。


SMA在多学科综合管理、疾病修正治疗药物等方面取得长足进步,明显提升了患者生存期及生活质量。然而,对于年龄较大的青少年与成人患者尚缺乏系统性临床诊疗指南规范指导临床工作。基于循证医学原则,来自全国多家SMA诊疗中心的多学科专家经过充分讨论,达成一致意见,为SMA临床规范化诊疗提供重要依据。
SMA为常染色体隐性遗传,通常父母双方为携带者,新发突变少,通常无核心家系同胞兄妹以外的家族史。SMA主要临床表现为对称性肌肉无力萎缩、肌张力下降。婴儿起病类型,常出生后即有软婴表现,运动发育明显迟滞,无法获得坐、站等运动里程碑,此后出现运动功能倒退、呼吸肌无力等,多夭折。幼儿起病者可出现运动发育迟缓,常可短暂获得独坐、站立等功能,随后丧失这些运动功能,出现脊柱侧凸、关节畸形和呼吸衰竭等。
儿童青少年起病类型则通常可获得重要运动里程碑,但之后出现从下肢近端起始,向上肢近端、四肢远端和头颈部发展的进行性肌肉无力萎缩,运动功能逐渐丧失,晚期合并脊柱侧凸、关节挛缩、张口受限、呼吸困难等。
青少年、成人SMA患者表现为隐匿起病、缓慢进展的肢体无力萎缩,从下肢近端起始,以蹲起、上楼能力下降为初始表现,后向上肢近端、上下肢远端、躯干肌群、呼吸肌及球部肌肉进展。病程常达数年,甚至数十年。部分患者可合并肉跳感,早期多无吞咽困难、言语不清等症状,可伴轻度脊柱侧弯及关节挛缩。青少年、成人SMA患者生长发育方面,虽运动发育略落后,但基本可获得全部运动里程碑。智能正常,无感觉异常、尿便障碍。少部分患者家庭有常染色体隐性遗传家族史。
SMA的检查方法
-
血清肌酸激酶检测:血清肌酸激酶水平正常或轻中度升高。
-
肌电图检测:提示广泛神经源性损害。
-
基因检测:MLPA , qPCR等可检测SMNl基因拷贝数变异,如第7号或者第7,8号外显子纯合缺失突变;巢式PCR可以检测SMNl基因外显子和邻近内含子区域的微小变异;二代测序可用于SMA鉴别诊断筛查其他肌无力相关疾病;三代时检测SMNl基因拷贝数变异和微小变异。

-
推荐意见:SMA起病年龄与病情进展差异大,建议在既往国际公认的SMA分型基础上,针对青少年及成人SMA患者给出更为清晰的分型标准(n级推荐,D级证据)。修订版新分型有待在今后临床实践中不断检验。
-
推荐意见:临床诊断下运动神经元综合征,符合下肤近端起病特点,需考虑SMA。采用靶向突变分析进行分子遗传学诊断,若确认SMNl基因7号外显子纯合缺失可确诊SMA。若SMNl基因杂合缺失,则需进一步行特殊Sanger测序,明确未缺失SMNl基因是否存在致病性微小突变(I级推荐,C级证据);
-
推荐意见:诺西那生的多个随机对照临床试验及真实世界研究结果、荟苹分析,证实其用于SMA治疗的有效性和安全性(I级推荐,A级证据);
-
推荐意见:利司扑兰拥有年龄跨度大及不同疾病状态患者的高质量随机对照临床试验结果,且其中包含中国SMA患者数据,初步证实了有效性和安全性(I级推荐,B级证据)。
-
推荐意见:SMA患者孕育后代时,应对其配偶进行携带者检测,若其配偶亦为携带者,则应进行产前诊断(I级推荐,C级证据)。若夫妇双方均携带SMNl基因突变,需进行产前诊断(I级推荐,C级证据)。

